
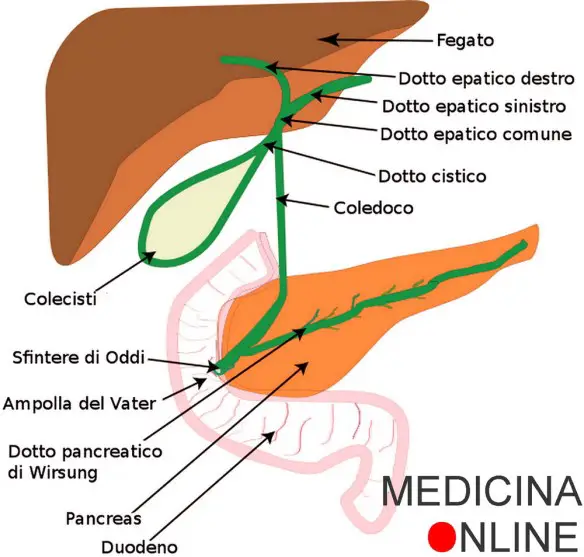
 La colangite biliare primitiva (anche chiamata cirrosi biliare primitiva, CBP) è una malattia cronica, a verosimile patogenesi autoimmune, che colpisce i dotti biliari di piccolo e medio calibro. Determina così ristagno cronico di bile (colestasi) che porta a fibrosi (formazione di tessuto cicatriziale) e cirrosi epatica. La malattia colpisce prevalentemente donne (rapporto donne-uomini pari a 9:1) tra i 40 e 60 anni di età. In Italia la CBP colpisce circa 20 persone ogni 100.000 abitanti.
La colangite biliare primitiva (anche chiamata cirrosi biliare primitiva, CBP) è una malattia cronica, a verosimile patogenesi autoimmune, che colpisce i dotti biliari di piccolo e medio calibro. Determina così ristagno cronico di bile (colestasi) che porta a fibrosi (formazione di tessuto cicatriziale) e cirrosi epatica. La malattia colpisce prevalentemente donne (rapporto donne-uomini pari a 9:1) tra i 40 e 60 anni di età. In Italia la CBP colpisce circa 20 persone ogni 100.000 abitanti.
Cause
La precisa causa di tale patologia non è nota; sembrerebbero implicati fattori genetici, legati ad una disfunzione del sistema immunitario, e fattori ambientali, come l’interazione dell’organismo con alcuni agenti infettivi.
Sintomi
I sintomi cardinali sono il prurito e la stanchezza. Il prurito, generalmente diffuso a tutto il corpo, è presente nella maggior parte dei pazienti ed ha intensità variabile e fluttuante nel tempo. L’astenia, non correlata con la gravità della malattia, può manifestarsi anche molto precocemente nel decorso di malattia. Si può presentare come affaticamento, difficoltà a concentrarsi, stato depressivo o eccessiva sonnolenza diurna.
Decorso
La malattia ha un andamento cronico e solitamente progredisce in modo lento e graduale, con periodi anche lunghi di relativo benessere. Solo una minor parte dei pazienti presenta alla diagnosi la cirrosi, il quadro avanzato della malattia epatica, o progredisce verso di essa. In questo stadio la fibrosi interessa tutto il fegato comportandone il suo corretto funzionamento.
Diagnosi
Nella metà dei casi la PBC viene diagnosticata in modo casuale quando, per altri accertamenti o durante uno screening, vengono rilevati anomali livelli dei marcatori di patologia epatica: le transaminasi (AST e ALT) e soprattutto gli indici di colestasi (gamma-GT e fosfatasi alcalina).
La diagnosi è data dalla presenza di almeno due dei seguenti tre criteri:
- positività della ricerca degli Anticorpi Anti-Mitocondrio (AMA) (che si riscontra nel 95% dei casi) a titolo adeguato (1:40 all’immunofluorescenza indiretta);
- persistenza per oltre 6 mesi di alti valori di fosfatasi alcalina (maggiori di 1.5 volte);
- biopsia epatica compatibile.
Gli AMA sono considerati anticorpi altamente specifici e sensibili, e la loro presenza nel siero è virtualmente diagnostica. Talora si rileva anche la presenza degli Anticorpi Anti-Nucleo (ANA), soprattutto nei casi in cui siano assenti gli AMA (condizione chiamata “Colangite Autoimmune”).
La biopsia epatica dà la certezza diagnostica e mostra una “colangite cronica destruente non suppurativa” (ovvero la presenza di granulomi che danneggiano i dotti biliari). Il danno istologico è classificato in 4 stadi, solo l’ultimo dei quali corrisponde ad una franca cirrosi:
- stadio 1: infiltrato infiammatorio portale;
- stadio 2: infiammazione e/o fibrosi periportale con proliferazione dei piccoli dotti biliari;
- stadio 3: setti fibrosi;
- stadio 4: cirrosi biliare (noduli di rigenerazione).
Malattie associate e complicanze
Rilevante è la possibilità che alla PBC si associno altre malattie autoimmuni, per questo è consigliabile effettuare uno screening sierologico per rilevarne la presenza.
- fino al 70% dei pazienti lamenta i sintomi della sindrome di Sjögren (caratterizzata da secchezza orale ed oculare e da artralgie);
- frequente è anche la tiroidite di Hashimoto, che può presentare i sintomi dell’ipotiroidismo (stanchezza, torpore, aumento ponderale);
- meno comuni sono l’epatite autoimmune (sindrome overlap), la celiachia ed il diabete mellito di tipo 1;
- più rare sono l’artrite reumatoide, la porpora trombocitopenica idiopatica, la sclerodermia, e la glomerulonefrite membranosa.
L’osteoporosi è piuttosto frequente in quanto:
- i pazienti sono frequentemente donne e con età superiore a 50 anni, quindi sono spesso affette da osteoporosi post-menopausale;
- la stasi biliare non consente un corretto assorbimento intestinale della vitamina D esogena (che è liposolubile);
- il danno epatico determina una ridotta attivazione della vitamina D endogena.
È inoltre aumentato il rischio di sviluppare l’epatocarcinoma, per il quale è consigliabile intraprendere un programma di sorveglianza (ecografia epatica e dosaggio dell’alfa-fetoproteina). Nelle fasi avanzate della malattia compaiono i segni dell’insufficienza epatica e dell’ipertensione portale:
- ittero;
- varici esofagee;
- ascite;
- edemi declivi;
- encefalopatia epatica;
- turbe della coagulazione (emorragie)
Terapia
Innanzitutto possono giovare alcune norme igieniche, finalizzate a preservare il più possibile la funzionalità epatica residua:
- astensione dal consumo di alcol
- mantenimento di un corretto peso corporeo (indice di massa corporea <25 kg/m2)
- seguire una dieta iposodica (per prevenire l’ipertensione portale)
- limitare l’uso di farmaci epatotossici (es. FANS)
- valutare la possibile vaccinazione contro i virus dell’epatite A e B
La terapia farmacologica della PBC si basa elettivamente sull’assunzione dell’Acido UrsoDesossiColico (UDCA) ad alto dosaggio (da 15 fino a 25 mg/Kg/die), che ha potere coleretico ed antinfiammatorio. Questo farmaco determina, nella maggior parte dei casi, una rapida diminuzione dei marcatori patologici, ed è, secondo numerosi studi clinici, in grado di rallentare la progressione della malattia e quindi di aumentare la sopravvivenza. L’UDCA è ben tollerato dall’organismo ed è sostanzialmente privo di effetti collaterali (raramente diarrea). Solo nei casi in cui il prurito sia persistente viene prescritta la Colestiramina, una resina che neutralizza gli acidi biliari ritenuti dall’organismo (responsabili di tale sintomo). Farmaci ad attività immunosoppressiva (come metotrexato, budesonide e colchicina) sono stati sperimentati vista la natura autoimmune della malattia, ma i risultati non si sono rivelati soddisfacenti, pertanto non vengono normalmente utilizzati. Vi si ricorre qualora la risposta all’UDCA (valutata in base all’abbattimento degli indici biochimici come la fosfatasi alcalina) risulti insufficiente. Alcuni studi clinici hanno mostrato risultati incoraggianti da parte del Bezafibrato, un farmaco ipolipemizzante con attività epatoprotettiva. Recentemente l’acido Obeticolico (agonista del recettore nucleare FXR, che regola la sintesi ed il trasporto degli acidi biliari) è stato registrato come farmaco di seconda linea nei paziente con risposta insoddisfacente all’UDCA o intolleranti all’UDCA. Per l’osteoporosi si procede alla supplementazione di vitamina D (preferibilmente la forma 25-idrossilata o calcifediolo) e calcio; eventualmente si può ricorrere a farmaci anti-fratturativi come i bisfosfonati (es. alendronato 70 mg/settimana). L’evoluzione allo stadio terminale (cirrosi, ipertensione portale, insufficienza epatica) richiede inevitabilmente il trapianto di fegato, proprio per il quale la PBC rappresenta una delle principali indicazioni. La ricorrenza della malattia sul trapianto è bassa e la percentuale di sopravvivenza ottima.
Prognosi e aspettativa di vita
I principali fattori prognostici sono:
- la precocità della diagnosi
- l’età alla diagnosi
- lo stadio istopatologico rivelato dalla biopsia
- l’entità della sintomatologia
- la positività a specifici tipi di ANA (anticorpi anti-nucleo), come gli anti-gp210, anti-sp100, anti-sp140 e anti-p62
- la risposta alla terapia (in particolare la diminuzione della fosfatasi alcalina ed i livelli di bilirubina)
- la sovrapposizione con altre patologie autoimmuni
La prognosi a medio-lungo termine (fino a 15 anni) può essere calcolata usando 2 score prognostici recentemente sviluppati: il GLOBE score ed il UK-PBC risck score. La CBP ha progressione estremamente variabile. I pazienti asintomatici hanno una aspettativa di vita superiore ai pazienti sintomatici ma significativamente inferiore rispetto a controlli sani di pari sesso ed età. La sopravvivenza media dei pazienti asintomatici è risultata variare tra 10 e 16 anni in due studi con un follow-up fino a 24 anni. I 2/3 dei pazienti asintomatici alla prima presentazione sviluppano sintomi (fatica, prurito, ittero, ascite, emorragia da varici esofagee od encefalopatia) nell’arco di 2-4 anni, mentre 1/3 dei pazienti può rimanere senza sintomi anche fino a 10 anni. La sopravvivenza media dei
pazienti sintomatici è di circa 7 anni. Non è stato identificato alcun parametro che consenta di prevedere lo sviluppo dei sintomi, nè che sia di valore prognostico nel paziente asintomatico. L’associazione con malattie autoimmuni è indicativa di prognosi meno favorevole. Nei pazienti con malattia avanzata, uno dei principali obiettivi del medico è quello di decidere la giusta tempistica per ricorrere al trapianto di fegato. A tale scopo sono stati sviluppati diversi modelli prognostici, basati su analisi di
regressione multipla (Cox), i quali prendono in considerazione combinazioni di più variabili.
Leggi anche:
- Insufficienza epatica lieve, acuta e cronica: dieta e rischio di morte
- Cirrosi epatica e fegato: sintomi, dieta, diagnosi, terapia e prevenzione
- Funzionalità epatica; cos’è, cosa indica e come si misura
- Foetor hepaticus: causa, odore, definizione, significato
- Epatiti croniche: cosa sono, sintomi, diagnosi e cura
- Transaminasi alte, basse, cosa sono, cosa indicano e come si curano
- Vena porta e sistema portale: anatomia e funzioni della circolazione epatica
- Legatura/sclerosi delle varici esofagee: perché si esegue, quali sono i rischi?
- Elastografia epatica (FibroScan) per cirrosi: valori, preparazione all’esame, risultati
- Calcolosi colecisti: sintomi, dieta e terapie dei calcoli biliari
- Colecistite acuta e cronica litiasica e alitiasica: cause, terapia, dieta e rimedi naturali
- Dipendenza da alcol: come fare per smettere di bere alcolici e superalcolici
- L’alcol è una droga?
- Dove si trova il fegato ed a che serve?
- Differenza tra cirrosi e fibrosi
- Bile: dove si trova, a che serve e da cosa è composta?
- Bilirubina diretta e indiretta: ittero, significato, patologie collegate
- Ittero emolitico, colestatico, ostruttivo, neonatale: significato, occhi, cura
- Pelle gialla: differenza tra ittero e carotenodermia
- Anemia emolitica: farmaci, bilirubina, ittero, diagnosi di laboratorio
- Fegato ed epatociti: anatomia, funzioni e patologie in sintesi
- L’esofagogastroduodenoscopia: cos’è, preparazione, è dolorosa o pericolosa?
- Colangiopancreatografia retrograda (ERCP): cos’è, preparazione, è dolorosa o pericolosa?
- Fegato ed epatociti: anatomia, funzioni e patologie in sintesi
- Cistifellea: cos’è, a cosa serve e dove si trova
- Dotto epatico comune, cistico e coledoco: anatomia del sistema biliare
- Duodeno: anatomia e funzioni in sintesi
- Pancreas: anatomia e funzioni in sintesi
- Cistifellea: cos’è, a cosa serve e dove si trova
- Si può vivere senza cistifellea?
- Riconoscere i differenti tipi di vomito a seconda del colore
- Vomito: le cause più frequenti
- Vomito: rimedi naturali e cure farmacologiche (farmaci anti-emetici)
- Sindrome post-colecistectomia: conseguenze dell’asportazione della cistifellea
- Differenza tra intestino tenue e crasso
- Mal di pancia e di stomaco: da cosa può dipendere e quali sono le cure
- Mal di pancia forte: quando chiamare il medico?
- Differenza tra ulcera gastrica, duodenale, peptica ed esofagea
- Ulcera peptica: complicanze, cura, dieta, quando è pericolosa
- Ecografia renale: esecuzione, indicazioni, costo
- Ecografia alla vescica vuota o piena: preparazione, costo, svolgimento
- Feci dalla bocca: il vomito fecaloide
- Incontinenza fecale: dieta, Alzheimer, parto, cause e terapie
- Il mio alito odora di feci: cause, quando è pericoloso e rimedi
- Morbo di Crohn: cos’è, cause scatenanti, sintomi, cure e dieta
- Differenze tra ileo meccanico ed ileo paralitico: cause, sintomi e trattamenti
- Colite ulcerosa: cause, diagnosi, cura, dieta, cosa mangiare, rimedi
- Differenza tra colon irritabile, colite e colite spastica: sono la stessa cosa?
Lo Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o unisciti al nostro gruppo Facebook o ancora seguici su Twitter, su Instagram o su Pinterest, grazie!
