
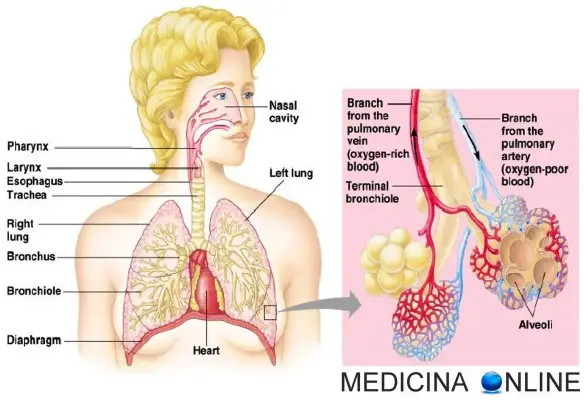
 La proteinosi polmonare alveolare (PAP) è una rara malattia a eziologia sconosciuta (le sue cause non sono quindi note), caratterizzata dal punto di vista anatomo-patologico dalla deposizione negli spazi alveolari di materiale granulare positivo all’acido periodico di Schiff (PAS), costituito in massima parte da fosfolipidi e proteine. La proteinosi polmonare alveolare insorge prevalentemente in uomini o donne in buona salute tra i 20 e i 60 anni. Occasionalmente, la PAP è comparsa in pazienti dopo esposizione a polveri inorganiche (p. es., silice, alluminio, titanio) e in pazienti affetti da infezioni croniche dovute allo Pneumocystis carinii, vari tumori ematologici, malattie mieloproliferative o immunosoppressione. Il significato di queste associazioni è incerto. Le alterazioni anatomo-patologiche sono limitate ai polmoni. Il rivestimento alveolare e le cellule interstiziali sono tipicamente normali, ma gli alveoli appaiono ripieni di granuli amorfi PAS-positivi contenenti varie proteine sieriche e non sieriche. La concentrazione dei lipidi negli spazi alveolari è elevata, forse a causa di un’alterazione della clearance dei fosfolipidi a livello alveolare. Raramente si ha fibrosi interstiziale. Il processo patologico può presentarsi diffuso o localizzato; interessa più frequentemente le regioni polmonari basali e posteriori ma in alcuni casi colpisce solamente i segmenti anteriori. La pleura e il mediastino non sono interessati
La proteinosi polmonare alveolare (PAP) è una rara malattia a eziologia sconosciuta (le sue cause non sono quindi note), caratterizzata dal punto di vista anatomo-patologico dalla deposizione negli spazi alveolari di materiale granulare positivo all’acido periodico di Schiff (PAS), costituito in massima parte da fosfolipidi e proteine. La proteinosi polmonare alveolare insorge prevalentemente in uomini o donne in buona salute tra i 20 e i 60 anni. Occasionalmente, la PAP è comparsa in pazienti dopo esposizione a polveri inorganiche (p. es., silice, alluminio, titanio) e in pazienti affetti da infezioni croniche dovute allo Pneumocystis carinii, vari tumori ematologici, malattie mieloproliferative o immunosoppressione. Il significato di queste associazioni è incerto. Le alterazioni anatomo-patologiche sono limitate ai polmoni. Il rivestimento alveolare e le cellule interstiziali sono tipicamente normali, ma gli alveoli appaiono ripieni di granuli amorfi PAS-positivi contenenti varie proteine sieriche e non sieriche. La concentrazione dei lipidi negli spazi alveolari è elevata, forse a causa di un’alterazione della clearance dei fosfolipidi a livello alveolare. Raramente si ha fibrosi interstiziale. Il processo patologico può presentarsi diffuso o localizzato; interessa più frequentemente le regioni polmonari basali e posteriori ma in alcuni casi colpisce solamente i segmenti anteriori. La pleura e il mediastino non sono interessati
Sintomi e segni
Il decorso naturale della PAP è ignoto e il quadro clinico varia enormemente. La condizione può progredire, rimanere stabile o guarire spontaneamente. Alcuni pazienti sono asintomatici; altri presentano grave insufficienza respiratoria. La maggior parte dei pazienti presenta dispnea da sforzo progressivamente ingravescente e tosse solitamente non produttiva. Coloro che fumano sigarette di solito producono espettorato. L’esame dell’escreato non fornisce alcun contributo. Raramente si sviluppano infezioni secondarie non batteriche (p. es., da Nocardia, Mycobacteria, Aspergillus e Cryptococcus sp). Sebbene il paziente sia febbrile all’esordio o subito dopo, una febbre persistente è rara a meno che non si verifichi un’infezione secondaria. I sintomi extrapolmonari sono insoliti. I reperti obiettivi sono limitati ai polmoni, ma possono essere assenti nonostante un interessamento parenchimale diffuso visibile alla rx del torace. Fini crepitii inspiratori sono in genere rilevabili sulle aree polmonari interessate.
Diagnosi
Una diagnosi specifica impone una biopsia polmonare o una broncoscopia con lavaggio broncoalveolare segmentale. Il liquido di lavaggio richiede speciali procedure di colorazione. Reperti caratteristici sono osservabili al microscopio ottico o elettronico nel tessuto e nel liquido di lavaggio. Le anormalità di laboratorio tipiche comprendono la policitemia, l’ipergammaglobulinemia e gli elevati livelli sierici di LDH. La rx torace mostra di solito un quadro di opacità a “farfalla” che ricorda quello dell’edema polmonare; il cuore appare normale. I linfonodi ilari non sono aumentati di volume. La TC ad alta risoluzione mostra opacità a vetro smerigliato e ispessimento delle strutture intralobulari e dei setti interolobulari con tipico aspetto poligonale, definito “pavimentazione pazza”. Sono in genere lievemente ridotti la capacità vitale, il volume residuo, la capacità residua funzionale, la capacità polmonare totale e la capacità di diffusione del monossido di carbonio misurata mediante respiro singolo. La disfunzione polmonare ostruttiva non è una caratteristica della malattia. L’ipossiemia può essere presente a riposo o, se la malattia è lieve, soltanto durante uno sforzo leggero o moderato. La PaO2 si mantiene di solito bassa respirando O2 al 100%, segno di uno shunt intrapolmonare destro-sinistro.
Prognosi e terapia
L’invalidità da insufficienza respiratoria è comune, ma raramente sopraggiunge la morte se la terapia con lavaggio broncopolmonare viene eseguita nei pazienti che ne hanno necessità. Le infezioni secondarie devono essere subito riconosciute e trattate. I pazienti asintomatici o con una sintomatologia minima non richiedono alcun trattamento, ma vanno tenuti in osservazione per le esacerbazioni che possono portare all’insufficienza respiratoria. Il trattamento è indicato solo per i pazienti con sintomi e ipossiemia significativi. Con il paziente in anestesia generale, i polmoni di solito vengono lavati uno per volta e a distanza di 3-5 gg l’uno dall’altro. La terapia più efficace è il lavaggio di un intero singolo polmone attraverso un tubo endotracheale a doppio lume, con ripetuti cicli di riempimento e di svuotamento, usando 1-2 l di soluzione riscaldata di NaCl 0,9%. Alcuni pazienti richiedono solamente un lavaggio perché i sintomi e gli infiltrati non recidivano; altri individui necessitano di lavaggi q 6 o 12 mesi per molti anni. Molti farmaci, compresi lo ioduro di potassio e gli enzimi proteolitici (p. es., la tripsina e la streptochinasi-streptodornasi), sono stati provati con successo variabile. I corticosteroidi per via sistemica sono stati usati senza successo e possono aumentare l’incidenza delle infezioni secondarie. Qualunque schema terapeutico è difficile da valutare poiché possono verificarsi remissioni spontanee e perché il numero di casi studiabili dai singoli ricercatori è limitato.
Leggi anche:
- Tumore al polmone: aspettativa di vita, sopravvivenza, fase terminale
- Tumore pleurico e mesotelioma: sopravvivenza e aspettativa di vita
- Cavo, liquido e versamento pleurico: fisiologia e patologia
- Differenza tra PM10 e PM2,5 e rispettivi effetti sulla salute
- Da oggi l’inquinamento dell’aria è ufficialmente un cancerogeno
- Polvere toracica: le case dei fumatori sono inquinate come Pechino
- Broncopolmonite: sintomi iniziali, contagio, prognosi, morte
- Polmonite ab ingestis: cause, tempi di guarigione, morte, sopravvivenza
- Tracheite virale, batterica, allergica: quanto dura e come si cura
- Laringite acuta e cronica: antibiotico, catarrale, cure, rimedi naturali
- Faringite virale e batterica: rimedi, quanto dura, è contagiosa?
- Bronchite: sintomi, durata, cura, rimedi naturali, è contagiosa?
- Mal di gola forte: rimedi naturali e farmaci per farlo passare
- Cordite in medicina: significato, da reflusso, sintomi, cura
- Laringoscopia diretta e indiretta: anestesia, costo, è dolorosa?
- Differenza tra laringoscopia diretta, indiretta, rigida, flessibile
- Intossicazione da monossido di carbonio: sintomi, danni permanenti, morte
- Atelettasia: significato, polmonare, cause, sintomi, cura, riabilitazione
- Laringospasmo: virale, da ansia, da stress, significato
- Broncospasmo in bambini e anziani: rimedi, quanto dura, paradosso
- Differenza tra laringospasmo e broncospasmo
- Morte per soffocamento: segni, sintomi, fasi e tempi
- Broncopneumopatia cronica ostruttiva (BPCO): sintomi, diagnosi e cura
- Differenze tra faringite, laringite e tracheite: vari tipi di mal di gola
- Asma bronchiale in bambini e adulti: cause, sintomi e cura
- Laringite ipoglottica (pseudocroup): cause, sintomi, cosa fare
- Croup nel bambino: significato, cause, sintomi, terapia, mortalità
- Tampone faringeo: preparazione, digiuno, positivo o negativo
- Differenza tra tonsillite, faringite, laringite, tracheite, adenoidite
- Differenza tra mal di gola e tonsillite
- Differenza tra mal di gola virale e batterico
- Abbassamento della voce: cause, rimedi, quando andare dal medico
- Bronchiolite in neonati e bambini: sintomi, cause, è pericolosa?
- Bronchiolite nei bimbi: mortalità, pericoli, complicazioni e durata
- Differenza tra croup e pseudocroup
- Differenza tra bronchite, bronchite asmatica e asma bronchiale
- Broncoscopia polmonare con biopsia: a cosa serve, fa male, è pericolosa?
- Polipi, noduli e granulomi delle corde vocali: cause, sintomi e cure
- Dove si trovano le corde vocali: anatomia e funzioni in sintesi
- Perché viene la tosse e come faccio a farla passare?
- Perché si starnutisce? Cosa fare se si continua a starnutire?
- Differenza tra tosse secca, grassa, cronica e con catarro
- Perché ci viene la febbre e perché non dobbiamo aver paura di lei
- Differenza tra raffreddore e influenza: sintomi comuni e diversi
- Come e quando abbassare la febbre? Farmaci e rimedi casalinghi
- Tonsille gonfie, infiammate e con placche rimedi in adulti e bambini
- Tonsillectomia: cosa mangiare, convalescenza, dolore, in adulti e bambini
- Tonsille palatine, linguali, faringee e tubariche: funzioni e patologia
- Differenza tra tonsille e adenoidi
- Adenoidi: ingrossate, operazione, convalescenza, ricrescono?
- Adenoidite acuta e cronica in bambini ed adulti: cause, sintomi, cure
- Differenze tra Paracetamolo, Tachipirina, Ibuprofene, Aspirina, Efferalgan e Co-Efferalgan
- Asfissia: sintomi, cure ed in quanto tempo si muore
- Respirazione artificiale bocca a bocca: quando farla e come farla
- Primo soccorso e BLS (Basic Life Support): cos’è e come si fa
- Cianosi in volto, mani o labbra: significato, cause, rischi, cure
- Saturazione dell’ossigeno: valori normali e patologici in anziani e bambini
- Cosa si prova a morire annegati, dissanguati, decapitati…
- Ipossiemia: significato, valori, sintomi, conseguenze, rischi, cure
- Ipercapnia: valori, terapia, conseguenze e trattamento
- Ipossia: valori, conseguenze, sintomi, cure
- Anossia: definizione, cause, sintomi, sinonimo, cure
- Ipocapnia: significato, cause, valori, alcalosi respiratoria
- Differenza tra ipossiemia e ipercapnia
- Differenza tra ipossiemia, ipossia, anossiemia ed anossia
- Differenza tra Mucosolvan e Fluimucil
- Spirometria diretta ed indiretta: come si esegue ed a cosa serve
- Sì al bagno dopo mangiato, ecco le vere 8 cause più frequenti di morte in acqua
- Croup nel bambino: significato, cause, sintomi, terapia, mortalità
- Differenza tra asfissia e soffocamento
- Differenza tra soffocamento e strangolamento
- Massaggio cardiaco: quando farlo e come farlo [LINEE GUIDA]
- Morte in culla (SIDS): prevenzione, cause, sintomi e percentuale dei casi
- Malattia da decompressione: terapia e fisiopatologia
- Embolia gassosa arteriosa da immersione: sintomi e cure
- Cosa succede al cibo nello stomaco dopo averlo ingerito?
- Apnea ostruttiva del sonno: cause, rischi, trattamenti e prevenzione
- Perché si russa e quali sono i rimedi per smettere di russare? I pericoli dell’apnea ostruttiva del sonno
- Crisi respiratoria acuta e rischio di morte: cosa fare?
- Pallore in viso: significato, sinonimo, cause, ansia, cosa mangiare
- Le 7 fasi della deglutizione (volontarie ed involontarie)
- Differenza tra disfagia di tipo ostruttivo e di tipo motorio
- Differenza tra disfagia ai liquidi e ai solidi
- Insufficienza respiratoria acuta e cronica: cause e conseguenze
- Intubazione: rischi, anestesia, rianimazione, dolore alla gola
- Tracheotomia possibilità di parlare, durata, conseguenze, quando si fa
- Tracheostomia: complicanze, parlare, percutanea, cibo e gestione
- Differenza tra tracheostomia percutanea e tradizionale (a cielo aperto)
- Differenza tra tracheotomia e tracheostomia
- Cricotiroidotomia: urgenza, complicanze, procedura ed indicazioni
- Differenza tra tracheotomia e cricotiroidotomia
- I migliori saturimetri professionali per uso ospedaliero e casalingo
- Come si misura la frequenza respiratoria?
- Frequenza respiratoria normale, alta, bassa, a riposo e sotto sforzo
- Differenze tra respiro normale e patologico
- Iperventilazione: significato, sintomi, alcalosi e conseguenze
- Dispnea ansiosa, notturna e cardiaca: sintomi, diagnosi e cura
- Differenza tra dispnea, apnea e tachipnea
- Enfisema polmonare: sintomi, tipi, cause, diagnosi e terapia
- Differenza tra insufficienza respiratoria di tipo 1 e 2
- Insufficienza polmonare lieve, severa, acuta: sintomi e cura
- I migliori elettrocardiografi ed Holter portatili
- I migliori sfigmomanometri e stetoscopi per misurare la pressione arteriosa
- I migliori glucometri di ultima generazione per misurare la glicemia
- I migliori misuratori di colesterolo LDL HDL e trigliceridi, affidabili e precisi
- Fenomeno di Raynaud: cause, sintomi e trattamento
- Cos’è l’edema, come e perché si forma?
- Pneumotorace spontaneo primario, secondario ed iperteso: cause, sintomi, terapie
- Tutti gli articoli sullo smettere di fumare
- Respiro patologico: le alterazioni del ritmo respiratorio normale
- Respiro di Biot ed apnee: caratteristiche e cause patologiche e non patologiche
- Respiro di Cheyne-Stokes: caratteristiche e cause patologiche e non patologiche
- Respiro di Falstaff: caratteristiche e cause
- Respiro di Kussmaul: caratteristiche e cause
- Idrotorace: cause, patologie, sintomi, diagnosi e cure
- Embolia polmonare: massiva, diagnosi, da tumore, terapia
- Polmoniti nosocomiali: cause, terapie e linee guida ATS
- Polmonite interstiziale, atipica, senza febbre: sintomi e cure in bimbi ed adulti
- Tumore al polmone operabile ed inoperabile: stadiazione
- Massaggio cardiaco: quante compressioni al minuto?
- Toracentesi: procedura, complicanze, rischi, è dolorosa?
- Parametri della spirometria: capacità, volumi, rapporti e flussi
- Empiema pleurico: guarigione, saccato, complicanze, RX, cura
- Differenza tra dispnea ed affanno
- Differenza tra polipnea e tachipnea
- Tipologie di respirazione nello yoga
- Apparato respiratorio: anatomia in sintesi, struttura e funzioni
- Trachea: anatomia e funzioni in sintesi
- Sistema nervoso: com’è fatto, a che serve e come funziona
- Differenza tra inspirazione e espirazione: l’atto respiratorio
- Differenza tra ventilazione polmonare e alveolare: spazio morto anatomico e fisiologico
Lo Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o unisciti al nostro gruppo Facebook o ancora seguici su Twitter, su Instagram o su Pinterest, grazie!
