
 Con il termine “facomatosi” (in inglese “phakomatoses”) o con “ectodermosi congenite”, in medicina si identifica un gruppo di malattie ereditarie che interessano la cute e altri organi, tra cui il cervello. In tale gruppo possiamo includere varie patologie, tra cui:
Con il termine “facomatosi” (in inglese “phakomatoses”) o con “ectodermosi congenite”, in medicina si identifica un gruppo di malattie ereditarie che interessano la cute e altri organi, tra cui il cervello. In tale gruppo possiamo includere varie patologie, tra cui:
- neurofibromatosi;
- sclerosi tuberosa;
- angiomatosi retino-cerebellare (malattia di Von Hippel-Lindau);
- angiomatosi cutanea associata ad anomalie del SNC (angiomatosi meningofacciale con calcificazione cerebrale o sindrome di Sturge-Weber);
- incontinentia pigmenti;
- sindrome di Bonnet – Dechaume – Blanc (sindrome di Wyburn-Mason);
- atassia-teleangectasia (sindrome di Louis-Bar).
La neurotibromatosi e la sclerosi tuberosa, due facomatosi tra le più diffuse, sono caratterizzate dalla presenza, nel sistema nervoso centrale (SNC), di formazioni benigne similtumorali (amartomi) che hanno la potenzialità di subire una trasformazione neoplastica.
In questo articolo ci occuperemo della sindrome di Von Hippel-Lindau.
Sindrome di Von Hippel-Lindau
La sindrome di Von Hippel-Lindau (VHL) è una sindrome a carattere ereditario autosomico dominante molto rara, caratterizzata dall’associazione di diverse forme di neoplasia, fra cui angiomi e altre forme di neoplasia del rene e feocromocitomi. Il nome della sindrome è dovuto a coloro che per primi la descrissero: l’oftalmologo tedesco Eugen von Hippel e il patologo svedese Arvid Lindau.
Sinonimi
La sindrome di Von Hippel-Lindau viene anche chiamata:
- angiomatosi retino-cerebellare;
- malattia di Von Hippel-Lindau;
- angiomatosi cerebello-retinica familiare;
- angiomatosi cerebelloretinica familiare;
- malattia di Hippel-Lindau;
- malattia di Lindau.
Epidemiologia
La prevalenza è di 1/53.000 casi; l’incidenza annuale è 1/36.000. I due sessi sono interessati in eguale misura. L’età media alla diagnosi è 26 anni, ma può manifestarsi a qualunque età.
Cause
La causa di questa malattia è una mutazione – trasmessa dai genitori ai figli – di un gene localizzato sul braccio corto del terzo cromosoma, mappato in 3p25-26, il quale codifica per la proteina VHL. Quest’ultima normalmente collabora con le elonghine B e C per legarsi ai fattori di trascrizione HIF (Hypoxia Induced Factor) idrossilati dalla presenza di ossigeno, ne provoca l’ubiquitinizzazione e la distruzione tramite il proteasoma. Il complesso coi geni HIF è responsabile delle risposte cellulari all’ipossia, fra cui lo sviluppo di neoangiogenesi e l’induzione della proliferazione cellulare; quindi VHL si comporta come un oncosoppressore e sono facilmente deducibili le conseguenze cliniche della sua mancanza. Il prodotto genico può essere totalmente assente (delezioni o mutazioni frameshift) con espressione anomala di codoni di STOP) oppure inattivo, portando a quadri clinici differenti, inoltre blocca il VEGF.
Trasmissione
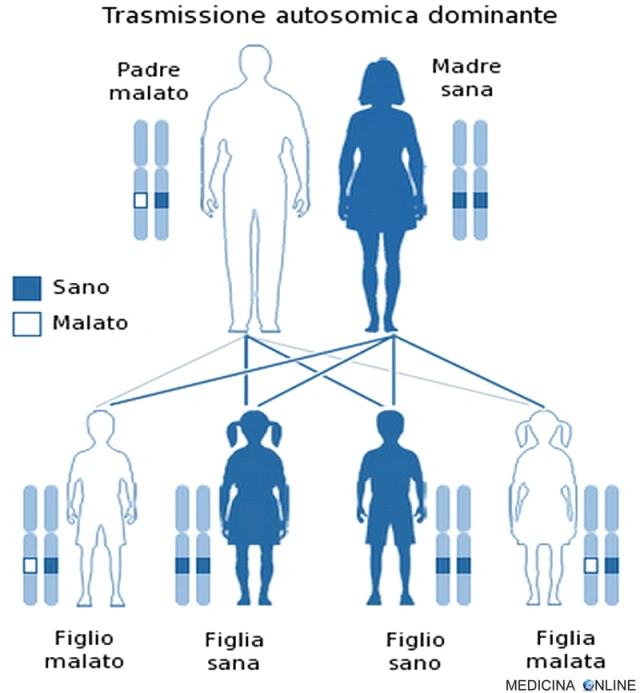
La sindrome di Von Hippel-Lindau viene trasmessa con modalità autosomico dominante. Una malattia è detta a trasmissione autosomica dominante quando basta una singola copia dell’allele difettoso per far sì che la malattia si esprima, a prescindere dal sesso (basta un solo genitore malato). Il figlio di un individuo affetto ha la probabilità del 50% di essere affetto, cioè 1 figlio su 2 è malato e può trasmettere a sua volta la malattia alla metà dei suoi figli. In questo caso non può esistere un “portatore sano” (cosa che invece si può verificare nella trasmissione autosomica recessiva): chi possiede l’allele alterato, ha la patologia, mentre chi non lo possiede è sano. Di conseguenza da due genitori sani nascono il 100% di figli sani, mentre se entrambi i genitori sono malati allora si avranno il 100% di figli malati.
Sintomi e segni
Il segno più comune sono gli emangioblastomi della retina (multipli e bilaterali in circa il 50% dei casi). Di solito sono asintomatici, ma possono causare distacco della retina, edema della macula, glaucoma e perdita della vista. Gli emangioblastomi del sistema nervoso centrale (SNC) sono presenti in circa il 40% dei casi e nel complesso nel 60-70% dei pazienti. Si localizzano spesso nel cervelletto, ma possono interessare anche il tronco encefalico e il midollo spinale. Sono benigni, ma possono essere sintomatici, a causa della compressione del tessuto nervoso adiacente. Nel cervelletto, si associano spesso ad aumento della pressione intracranica, cefalee, vomito, atassia del tronco e degli arti. Sono molto comuni le cisti renali multiple e la mortalità da RCC è molto elevata (70% dei casi). Alcuni pazienti sono affetti da feocromocitomi, che possono essere asintomatici, ma causare ipertensione. Sono inoltre presenti cisti e cistoadenomi epididimali e cisti pancreatiche multiple (nella maggior parte dei pazienti) e, in un limitato gruppo di casi (circa il 10%), tumori del sacco endolinfatico (ELST). Sono rari i paragangliomi della testa e del collo (0,5%). L’età media alla diagnosi dei tumori nella VHL è molto più giovane rispetto a quella descritta negli stessi tumori sporadici. È presente marcata variabilità intrafamiliare.
Clissificazione
A seconda del genotipo del soggetto sono rinvenibili quadri clinici differenti; in particolare la malattia è suddivisibile in 2 tipi. Il tipo 1 con assenza di feocromocitoma e il tipo 2 che invece lo manifesta. Nel primo caso la proteina spesso è mutata, ma presente; nel secondo caso invece è deleta. Inoltre il tipo 2 è diviso ulteriormente in 3 sottotipi: A, B e C (il primo con tumore a cellule renali, il secondo con anche emangioblastoma e il terzo con solo feocromocitoma). Sono anche riscontrati tumori endocrini (specie pancreatici), cisti renali (dovute all’edema da neoangiogenesi) e altre manifestazioni neoplastiche.
Diagnosi
La diagnosi è posta in presenza di un singolo tumore caratteristico (emangioblastoma della retina o del SNC, oppure RCC) e di una storia familiare positiva per la VHL. In assenza di una storia familiare (circa il 20% dei casi insorge de novo), è suggestiva la presenza di tumori multipli (due emagioblastomi o un emangioblastoma e un RCC). L’emocromo completo, la misurazione dei metaboliti delle catecolamine urinarie, l’esame delle urine e la citologia urinaria possono rivelare rispettivamente policitemia, presenza di feocromocitoma, anomalie urinarie e RCC. Gli studi di imaging sono utili per identificare i tumori renali, del SNC, del sacco endolinfatico, il feocromocitoma, le cisti renali e pancreatiche. La diagnosi differenziale si pongono con la neoplasia endocrina multipla, la neurofibromatosi, la malattia renale policistica, la sclerosi tuberosa, la sindrome di Birt-Hogg-Dubé e il feocromocitoma-paraganglioma ereditario, legati a mutazioni delle subunità della succinato deidrogenasi (SDHB, SDHC e SDHD). La diagnosi prenatale è possibile con l’analisi molecolare sugli amniociti o sui villi coriali, se la mutazione patogenetica è già nota in un familiare. Deve essere offerta la consulenza genetica.
Terapia
Si procede a intervento chirurgico, che viene effettuato se sono presenti lesioni spinali sintomatiche, lesioni cerebrali sintomatiche o in rapida evoluzione, neoplasie solide renali > 3 cm e/o neoplasie solide pancreatiche > 3 cm.
Prognosi
Leggi anche:
- Visita neurologica: svolgimento, esami, patologie, quando è necessaria?
- Sclerosi multipla: cause, sintomi, diagnosi e prognosi
- Sclerosi laterale amiotrofica (SLA): cause, sintomi, diagnosi e prognosi
- Test di Romberg: cos’è, a che serve, come si esegue
- Differenze tra sclerosi laterale amiotrofica e sclerosi multipla
- Atrofia muscolare progressiva: cause, sintomi, cura, aspettativa di vita
- Atrofia muscolare spinale: sintomi, trasmissione, tipi e cure
- Differenze tra atrofia muscolare progressiva e sclerosi laterale amiotrofica
- Riflesso di Babinski positivo: sintomi, diagnosi, come evocarlo
- Segno di Babinski positivo nel neonato e nel bambino: che significa?
- Segno di Babinski nella sclerosi multipla e nella SLA
- Segno di Brudzinski positivo e negativo: semeiotica nella meningite
- Segno di Hoffman positivo in SLA e sclerosi multipla
- Segno di Kernig positivo e negativo: semeiotica nella meningite
- Segno di Lasègue positivo e negativo in semeiotica
- Segno di Wasserman (Lasègue inverso) positivo in semeiotica
- Segno di Binda: cos’è e come si esegue
- Segno di Amoss (o del tripode): cos’è e come si esegue
- Segno di Magnus-De Klein: cos’è e come si esegue
- Malattia di Huntington: cos’è, ereditarietà, come si trasmette, età di insorgenza
- Differenza tra tremori, spasmi, miotonia, crampi, fascicolazioni, tic
- Differenza tra discinesia, ipocinesia, ipercinesia, tardiva e primaria
- Fascicolazioni muscolari, il tremolio spontaneo di un muscolo: cause e cure
- Differenza tra astenia, ipostenia, miastenia, ipotonia, nevrastenia, iperstenia, ipertonia
- Astenia, quando mancano le forze fisiche o mentali: cause, diagnosi, cure
- Nevrastenia (esaurimento nervoso): cause, diagnosi, cure
- Ipostenia (miastenia): cause, sintomi, diagnosi, terapie, complicanze, prognosi
- Ipotonia e atonia: definizione, etimologia, significato, esempi
- Ipotonia muscolare in neonati, adulti, anziani: cause, sintomi, cure, consigli
- Ipotonia nel lattante: cause, sintomi, diagnosi e trattamento
- Iperstenia: definizione, significato, etimologia, fisiologica e patologica
- Ipertonia: definizione, significato, etimologia
- Ipertonia muscolare: cause, tipi, sintomi, diagnosi, terapie
- Cos’è il tono muscolare, a che serve, come mantenerlo?
- Distonia focale e generalizzata: tipi, sintomi, diagnosi e terapie
- Miotonia: cause, sintomi, diagnosi e terapie
- Demenza senile: cause, sintomi, decorso e cure
- Demenza da corpi di Lewy: cause, decorso, Parkinson, aspettativa di vita
- Differenza tra morbo di Alzheimer, demenza senile, vascolare e reversibile
- Mielite: infettiva, cervicale, dorsale, trasversa, si guarisce?
- Meningite: contagio, sintomi, vaccino, gravità e profilassi
- Segni meningei e irritazione meningi in bambini ed adulti
- Meningite: incubazione, fulminante, sintomi, contagio e cura
- Encefalite: conseguenze, è contagiosa, danni, si guarisce?
- Encefalite autoimmune da anticorpi anti-NMDA: trattamento, sintomi
- Meningismo: triade, segni, cause, diagnosi, definizione e cura
- Differenza tra meningite e meningismo: qual è più grave?
- Differenza tra meningite, encefalite, meningoencefalite, encefalomielite
- Differenza tra meningite virale e batterica
- Differenza tra virus e batteri: chi è più pericoloso? Diagnosi, sintomi e terapia
- Puntura lombare: complicanze, risultati, è dolorosa, a che serve?
- Meningi: anatomia, funzioni e patologia in sintesi
- Liquido cefalorachidiano: dove si trova, perdita dal naso, prelievo
- Mielopatia: significato, tipi, sintomi, diagnosi,cure, consigli, prognosi
- Segni meningei e irritazione meningi in bambini ed adulti
- Tumore al cervello: cause, sintomi iniziali e tardivi, diagnosi, cura, sopravvivenza, aspettativa di vita
- Ipertensione endocranica: valori, cause, bradicardia, terapie
- Pseudotumor cerebri (ipertensione endocranica benigna) cause e cure
- Operato al cervello per un tumore mentre suona il clarinetto
- Tumore al cervello: operato mentre suona la chitarra e canta Yesterday
- Esoftalmo bi- e monolaterale: cause, gravità, conseguenze e cure
- Transilluminazione della testa di un neonato per la diagnosi di idrocefalo
- Idrocefalo: cause, terapia, conseguenze, aspettativa di vita
- Idrocefalo nel feto e neonatale: conseguenze e cura
- Pressione intracranica e pressione di perfusione cerebrale
- Sindrome della sella vuota: cause, sintomi, diagnosi e terapie
- Differenza idrocefalo iperteso, normoteso, comunicante, ostruttivo
- Macrocefalo in neonato e bambino: sintomi, cure e ritardo psicomotorio
- Shunt cerebrale e intervento per il drenaggio permanente
- Ventricoli cerebrali: anatomia e funzioni in sintesi
- Emorragia cerebrale da caduta e trauma cranico: sintomi, diagnosi e cure
- Emorragia cerebrale: non operabile, coma, morte, si può guarire?
- Emorragia cerebrale: operazione e tempi di riassorbimento
- Emorragia cerebrale: conseguenze, riabilitazione e recupero
- Emorragia subaracnoidea: cause, conseguenze, linee guida
- Ematoma subdurale: cos’è, da quale malattia è provocata, recidivo, decorso
- Coma da emorragia cerebrale: quanto può durare?
- Differenza tra proencefalo, mesencefalo, romboencefalo, telencefalo, diencefalo
- Differenza tra metencefalo e mielencefalo
- Emicrania con aura: cause, sintomi, diagnosi e trattamenti
- Emicrania senza aura: cause, sintomi, diagnosi e trattamenti
- Differenza tra emicrania con aura ed emicrania senza aura
- Cos’è l’aura emicranica?
- Ictus cerebrale emorragico e ischemico: cause, sintomi, diagnosi, cure, rischi
- Differenza tra ictus cerebrale ed attacco ischemico transitorio (TIA)
- Che cos’è un attacco ischemico transitorio (TIA)? Impara a riconoscerlo e potrai salvare una vita, anche la tua
- Aneurisma cerebrale rotto e non rotto: cause, sintomi, diagnosi e cura
- Cosa si prova e cosa succede quando si rompe un aneurisma cerebrale?
- Ictus, emorragia cerebrale cerebrale e TIA: cosa fare e cosa assolutamente NON fare
- Sindrome frontale o ipofrontalità: cause, sintomi, conseguenze, diagnosi, cure
- Malformazioni artero-venose cerebrali: sintomi e cura
- Ischemia: cos’è, cause, conseguenze, rischi, cure
- Necrosi: significato, definizione, sinonimo, cause, cure
- Trombo: cause, classificazione, trombosi venose, arteriose e sistemiche
- Infarto, ischemia, necrosi, aterosclerosi, trombo, embolo, ictus, miocardio… Facciamo chiarezza
- Tronco cerebrale (mesencefalo, ponte e bulbo) anatomia e funzioni in sintesi
- Morbo di Parkinson: cause, sintomi, decorso, terapie
- Morbo di Alzheimer: cause, sintomi, decorso, terapie
- Parestesie: significato, cause, rischi, diagnosi, cure, rimedi, esercizi
- Epilessia infantile ed in adulti: cause, sintomi, diagnosi, cosa fare
- Epilessia: come riconoscere un attacco e soccorrere un ammalato
- Differenza tra epilessia e convulsioni
- Differenza tra epilessia e sincope
- Differenza tra epilessia parziale e generalizzata
- Epilessia: riconoscere in tempo l’arrivo di una crisi e come comportarsi
- Epilessia infantile: come comportarsi col proprio figlio?
- Esame della sensibilità tattile, dolorifica, termica, vibratoria in neurologia
- Esame delle funzioni motorie e dei riflessi in neurologia
- Esame delle funzioni cerebrali superiori (corticali) in neurologia
- Asterissi (asterixis) in neurologia: caratteristiche, significato, esecuzione
- Torcicollo spasmodico e spasmi linguali, facciali, oromandibolari, della mano (distonie focali)
- Tic, ritmie, movimenti stereotipati, acatisia e trasalimento nel paziente neurologico
- Disturbi della stazione eretta e della deambulazione in neurologia
- Esame della motilità oculare e disturbi dei movimenti coniugati in neurologia
- Sordità, esame dell’udito e tecniche audiologiche speciali in neurologia
- Aprassia ideazionale, ideomotoria e melocinetica: sintomi, diagnosi, cure
- Aprassia: significato, etimologia, tipi, zone del cervello coinvolte
- Aprassia: test usati per la diagnosi e tipici errori aprassici
- Aprassia costruttiva (apractognosia): cause, sintomi, diagnosi, terapie
- Disartria: cause, sintomi, diagnosi, trattamento
- Perdita della coordinazione muscolare: l’atassia
- Differenze tra aprassia, atassia, disprassia, afasia, disartria, anartria
- Non riconoscere i volti dei propri cari: la prosopagnosia, cause, test e cure
- Spina bifida e difetti di chiusura del tubo neurale nel feto: trasmissione, prevenzione, diagnosi e cura
- Mielomeningocele, spina bifida e schisi vertebrale: complicanze, cura, prevenzione
- Encefalocele: cause, sintomi, diagnosi, cura e prevenzione
- Anche gli esseri umani possono avere una coda
- Sindrome di Arnold-Chiari: linguaggio, aspettative di vita, invalidità, mortalità
- Siringomielia (malattia di Morvan): cause, sintomi, diagnosi, cure
- Siringobulbia: cause, sintomi, diagnosi e trattamento
- Glasgow Coma Scale per la classificazione del coma
- Pediatric Glasgow Coma Scale in italiano: scala pediatrica del coma
- Differenza tra toracentesi, paracentesi e rachicentesi
- Narcolessia: cause, sintomi, cure e terapia farmacologica
- Catatonia: significato, definizione, cause, sinonimi e cure
- Cataplessia: causa, significato, nel sonno, cura ed etimologia
- Catalessia in medicina: cause, sintomi, nel sonno e cure
- Differenza tra catatonia, catalessia e cataplessia
- Paralisi del sonno e allucinazioni ipnagogiche: cause, pericoli, rimedi
- Morte cerebrale: diagnosi, sintomi, risveglio, durata, si può guarire?
- Coma: cause, risveglio, tipi, quanto dura, fasi, segni, irreversibile
- Differenza tra coma e coma farmacologico
- Elettroencefalogramma: preparazione, alterazioni, costo, rischi
- Sindrome locked-in: cause, riabilitazione, respirazione, cure
- Stato di minima coscienza: evoluzione, risveglio, riabilitazione
- Stato vegetativo: risveglio, riabilitazione, durata e caratteristiche
- Differenza tra stato vegetativo, di minima coscienza, coma, sonno e stato soporoso
- Differenza tra sindrome locked-in e stato vegetativo
- Differenza tra stato vegetativo persistente e permanente
- Differenza tra stato vegetativo e stato di minima coscienza
- Commozione cerebrale: cos’è, cosa fare, conseguenze, tempi di recupero
- Differenza tra commozione cerebrale, trauma cranico e contusione cerebrale
- Trauma cranico: ematoma, commotivo, sintomi tardivi, cosa fare
- Emorragia cerebrale: cause, sintomi premonitori, diagnosi e cura
- Differenza tra morte clinica, biologica, legale, apparente, improvvisa ed istantanea
- Poligono di Willis: anatomia e varianti anatomiche
- Emiplegia destra, sinistra, spastica, flaccida: significato e riabilitazione
- Emiparesi destra, sinistra, facciale e neonatale: cause, sintomi e cure
- Paraplegia: etimologia, significato, sintomi, cura e riabilitazione
- Tetraplegia: significato, cause, cure e riabilitazione
- Diplegia: definizione, cause e sintomi
- Differenza tra emiplegia, emiparesi, diplegia, paraplegia, tetraplegia
- Differenza emiparesi, diparesi, tetraparesi, monoparesi, triparesi
- Differenza tra paraplegia e diplegia
- Classificazione generale delle paresi e delle plegie
- Anosognosia e Sindrome neglect: significato, test e trattamento
- Sindrome neglect (negligenza spaziale unilaterale): cura e riabilitazione
- Sistema nervoso: com’è fatto, a che serve e come funziona
- Sistema nervoso simpatico: funzioni
- Sistema nervoso parasimpatico: funzioni
- Differenza tra afasia, disartria ed aprassia
- Area di Broca: funzioni ed afasia di Broca
- Area di Wernicke: funzioni ed afasia di Wernicke
- Differenza tra afasia di Broca e di Wernicke
- Cervelletto: anatomia esterna ed interna
- Cervelletto: le lesioni cerebellari più comuni
- Le funzioni del cervelletto: apprendimento e correzione dei movimenti del corpo
- Com’è fatto il cervello, a che serve e come funziona la memoria?
- Cervello maschile e femminile: quali sono le differenze?
- Sistema nervoso autonomo simpatico e parasimpatico: anatomia e funzioni
- Ipotalamo: anatomia, struttura e funzioni
- Differenze tra ipotalamo, ipofisi, neuroipofisi e adenoipofisi
- Differenza tra midollo osseo e spinale
- Differenza tra sistema nervoso centrale e periferico: anatomia e funzioni in sintesi
- A cosa serve il midollo osseo?
- Differenza tra midollo osseo e cellule staminali
- Differenza tra midollo spinale e allungato
- Differenza tra epifisi, diafisi, metafisi ed ipofisi
- Patologie di ipotalamo e ipofisi
- Ipofisi (ghiandola pituitaria): anatomia, funzioni e ormoni secreti
- Asse ipotalamo-ipofisario: fisiologia e ormoni rilasciati
- Si può morire di epilessia?
- Sindrome pseudobulbare: cause, sintomi, diagnosi e terapie
- Malattia di Binswanger: cause, sintomi, diagnosi, cure
- Sistema piramidale e fascio genicolato: anatomia, decorso, funzioni
- Sistema extrapiramidale: anatomia, decorso, vie, funzioni, patologie
- Differenza tra via piramidale ed extrapiramidale
- Miclono: cause, sintomi, caratteristiche, quando preoccuparsi, cure
- Spasmi muscolari e mioclonie: da cosa sono causati?
- Spasmi muscolari e mioclonie: cause, diagnosi e cura delle contrazioni involontarie
- Spasmi muscolari e mioclonie: cura, trattamento e rimedi
- Spasmi muscolari e mioclonie: come si fa la diagnosi?
Lo Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o unisciti al nostro gruppo Facebook o ancora seguici su Twitter, su Instagram o su Pinterest, grazie!
