
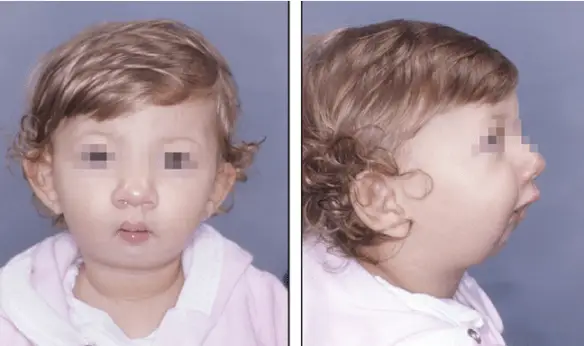
Micrognazia: la mandibola è di dimensioni ridotte
La sindrome di Beals (anche chiamata sindrome di Beals-Hecht o artrogriposi distale tipo 9 o aracnodattilia contrattuale congenita) è rara una sindrome che interessa il tessuto connettivo, caratterizzata da contratture multiple in flessione, aracnodattilia, cifoscoliosi grave, padiglioni auricolari abnormi e ipoplasia muscolare. Ricorda per molti versi un’altra sindrome, quella di Marfan, ma sembra che l’incidenza epidemiologica sia inferiore a quest’ultima (1 persona su 3000-5000 contro 1 persona su 25000-50000).
Differenza tra sindrome di Beals e di Marfan
Benché i segni clinici della sindrome di Beals siano in parte sovrapponibili a quelli associati alla sindrome di Marfan, la presenza di contratture articolari multiple (in particolare a livello dei gomiti, delle ginocchia e delle articolazioni delle dita), le orecchie ‘martellate’ e l’assenza di una dilatazione aortica significativa sono caratteristiche della sindrome di Beals, mentre sono piuttosto rare nella sindrome di Beals. In molti casi risulta comunque difficile una diagnosi differenziale su base clinica tra le due sindromi.
Età di esordio
Le prime anomalie della sindrome di Beals si riscontrano fra i primi giorni di vita del bambino e il compimento del primo anno.
Cause della sindrome di Beals
La causa della sindrome di Beals è un’anomalia genetica dell’FBN2, gene che codifica per la fibrillina-2, mappato sul cromosoma 5q3. La fibrillina-2 è una delle due forme omologhe di una proteina della matrice extracellulare. Anomalie nel gene FBN1, codificante per la fibrillina-1 e mappato sul cromosoma 15q21, determinano la sindrome di Marfan.
Leggi anche: Aracnodattilia, segno del pollice e del polso, sindrome di Marfan e di Beals
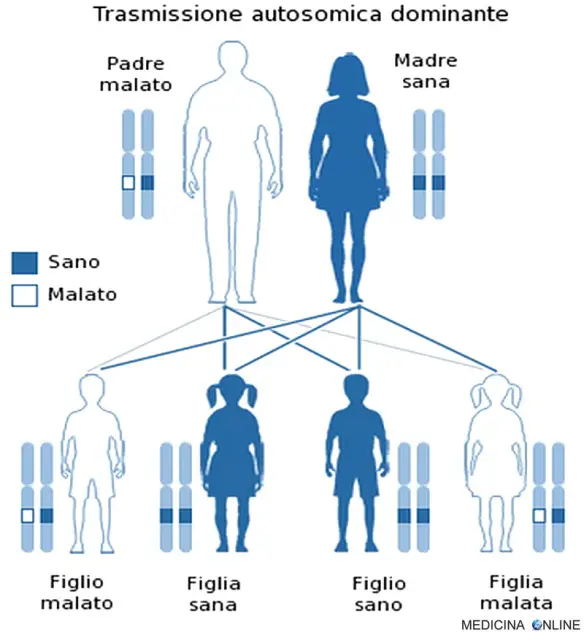
Trasmissione
La trasmissione della sindrome di Beals avviene con modalità autosomica dominante. Una malattia è detta a trasmissione autosomica dominante quando basta una singola copia dell’allele difettoso per far sì che la malattia si esprima, a prescindere dal sesso (basta un solo genitore malato). Il figlio di un individuo affetto ha la probabilità del 50% di essere affetto, cioè 1 figlio su 2 è malato e può trasmettere a sua volta la malattia alla metà dei suoi figli. In questo caso non può esistere un “portatore sano” (cosa che invece si può verificare nella trasmissione autosomica recessiva): chi possiede l’allele alterato, ha la patologia, mentre chi non lo possiede è sano. Di conseguenza da due genitori sani nascono il 100% di figli sani, mentre se entrambi i genitori sono malati allora si avranno il 100% di figli malati.
Segni e sintomi della sindrome di Beals
I sintomi e i segni clinici sono molteplici, si osservano: micrognazia (mandibola di dimensioni ridotte, vedi foto in alto), alta statura, il padiglione auricolare risulta abnorme e come se fosse accartocciato, cifoscoliosi, dilatazione aortica, contratture multiple in flessione, aracnodattilia ed ipoplasia muscolare.
Leggi anche: Donne nate senza vagina: è la sindrome di Rokitansky-Kuster-Hauser
Diagnosi della sindrome di Beals
Per la sindrome di Beals è possibile la diagnosi prenatale molecolare. L’ecografia consente di evidenziare le contratture articolari e l’ipocinesia, nei casi sospetti.
Terapie della sindrome di Beals
Non esistono attualmente terapie per curare a monte la malattia tuttavia esistono trattamenti atti a migliorare i segni clinici del paziente, come ad esempio la correzione chirurgica per la cifoscoliosi. È stato osservato un miglioramento spontaneo della camptodattilia e delle contratture, ma in ogni caso permane una camptodattilia residua. Fisioterapia e correzione chirurgica sono attualmente i trattamenti più comuni ed efficaci per contrastare la sindrome: se attuati precocemente riducono il rischio di complicanze in età adulta.
Prognosi ed aspettativa di vita nella sindrome di Beals
L’aspettativa di vita è pressoché normale, solo in rari casi essa viene ridotta a causa di gravi problemi cardiaci associati alla sindrome. E’ importante che il soggetto sia periodicamente controllato dal punto di vista cardiologico ed oftalmologico.
Leggi anche:
- Dita ippocratiche congenite e secondarie: cause, sintomi e terapie
- Quante ossa ci sono nella mano e come si chiamano?
- Sindrome di Marfan: trasmissione, sintomi e frequenza
- Microtia ed anotia: la malformazione congenita dell’orecchio
- Palatoschisi: cause, problemi derivanti e cure
- Labbro leporino (cheiloschisi): tipi, cause, problematiche e cure
- Arinia: la mancanza congenita del naso
- Le malattie genetiche più diffuse al mondo
- Sindrome di Klinefelter: cariotipo, cause, sintomi e cura
- Sindrome di Turner: cariotipo, cause, sintomi e segni caratteristici
- Sindrome di Down: cause, sintomi in gravidanza e nei neonati
- Atrofia muscolare progressiva: cause, sintomi, cura, aspettativa di vita
- Differenze tra sclerosi laterale amiotrofica e sclerosi multipla
- Morbo di Parkinson: cause, sintomi, decorso, terapie
- Morbo di Alzheimer: cause, sintomi, decorso, terapie
- Ectrodattilia: cause, cure ed immagini
- Polidattilia; cause, ereditarietà, sindromica e chirurgia
- Differenza tra polidattilia, sindattilia, ectrodattilia, oligodattilia e aracnodattilia
Dott. Emilio Alessio Loiacono
Medico Chirurgo
Direttore dello Staff di Medicina OnLine
