
 La causa per la maggior parte dei casi di Alzheimer è ancora in gran parte sconosciuta, ad eccezione che per casi dall’1% al 5% in cui sono state individuate le differenze genetiche esistenti.
La causa per la maggior parte dei casi di Alzheimer è ancora in gran parte sconosciuta, ad eccezione che per casi dall’1% al 5% in cui sono state individuate le differenze genetiche esistenti.
Diverse ipotesi cercano di spiegare la causa della malattia, che prendono in considerazione genetica, placche amiloidi e proteina Tau.
Leggi anche:
- Malattia di Parkinson: cause, sintomi, decorso, terapie
- Malattia di Alzheimer: cause, sintomi, decorso, terapie
Genetica
L’ereditabilità genetica della malattia di Alzheimer, sulla base di studi sui gemelli e familiari, comprendono dal 49% al 79% dei casi. Circa il 0,1% dei casi sono forme familiari autosomiche (non legati ai cromosomi sessuali) con ereditarietà dominante, che hanno un esordio prima dei 65 anni. Questa forma della malattia è conosciuta come Alzheimer giovanile. La maggior parte di questi casi, trasmissibili come caratteri mendeliani autosomici dominanti. possono essere attribuiti a mutazioni in uno dei tre geni: gene codificante il precursore dell’amiloide (APP 1) i geni codificanti per le Preseniline 1 (PS1) e 2(PS2). La maggior parte delle mutazioni nei geni APP e PS 1 e 2 aumentano la produzione di una piccola proteina chiamata Aβ42, che è la componente principale delle placche amiloidi senili. Alcune delle mutazioni alterano il rapporto tra Aβ42 e le altre principali forme come Aβ40 (non patologica) senza aumentare i livelli di Aβ42. Ciò suggerisce che le mutazioni della presenilina possono causare malattie anche se è inferiore la quantità totale di Aβ prodotto. Esistono varianti del gene APP che sono protettive.
La maggior parte dei casi di malattia di Alzheimer non presenta ereditarietà autosomica dominante e viene denominata AD sporadica, in cui le differenze ambientali e genetiche possono agire come fattori di rischio. Il fattore di rischio genetico più noto è l’eredità del allele ε4 della Apolipoproteina E (APO-E). Tra il 40 e il 80% delle persone con la malattia sono in possesso di almeno un allele APOEε4. L’allele APOEε4 aumenta il rischio della malattia di tre volte negli eterozigoti e di 15 volte negli omozigoti. Come molte malattie umane, effetti ambientali e modificatori genetici provocano penetranza incompleta. Ad esempio, alcune popolazioni della Nigeria non mostrano la relazione tra dose di APOEε4 e incidenza o età di insorgenza della malattia di Alzheimer, osservabile in altre popolazioni umane. Nei primi tentativi di screening, sono stati riconosciuti circa 400 geni associabili a AD sporadica con insorgenza tardiva (LOAD); ciò ha determinato una bassa resa dello screening. Più recenti studi di associazione sull’intero genoma (GWAS) hanno trovato 19 aree in geni che sembrano associate al rischio. Questi geni sono: CASS4, CELF1, FERMT2, HLA-DRB5, INPP5D, MEF2C, NME8, PTK2B, SORL1, ZCWPW1, SlC24A4, CLU, PICALM, CR1, BIN1, MS4A, ABCA7, EPHA1, CD2AP.
Mutazioni nel gene TREM2 sono state associate ad un rischio da 3 a 5 volte più elevato di sviluppare la malattia di Alzheimer..Si pensa che quando TREM2 è mutato ,i globuli bianchi nel cervello non sono più in grado di controllare la quantità di beta amiloide presenti.
Leggi anche:
- Demenza senile: cause, sintomi, decorso e cure
- Malattia di Alzheimer: sintomi delle fasi iniziali e tardive
Placche amiloidi
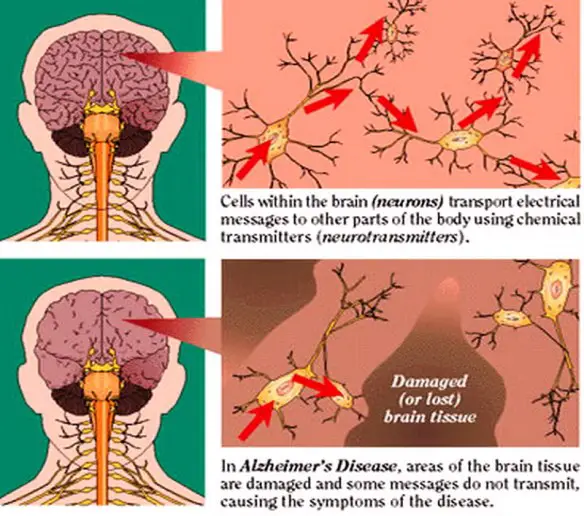
La malattia è dovuta a una diffusa distruzione di neuroni, principalmente attribuita alla beta-amiloide, una proteina che, depositandosi tra i neuroni, agisce come una sorta di collante, inglobando placche e grovigli “neurofibrillari”. La malattia è accompagnata da una forte diminuzione di acetilcolina nel cervello (si tratta di un neurotrasmettitore, ovvero di una molecola fondamentale per la comunicazione tra neuroni, e dunque per la memoria e ogni altra facoltà intellettiva). La conseguenza di queste modificazioni cerebrali è l’impossibilità per il neurone di trasmettere gli impulsi nervosi e quindi la morte dello stesso, con conseguente atrofia progressiva del cervello nel suo complesso. A livello neurologico macroscopico, la malattia è caratterizzata da una diminuzione nel peso e nel volume del cervello, dovuta ad atrofia corticale, visibile anche in un allargamento dei solchi e corrispondente appiattimento delle circonvoluzioni. A livello microscopico e cellulare, sono riscontrabili depauperamento neuronale, placche senili (dette anche placche amiloidi), ammassi neurofibrillari, angiopatia congofila (amiloidea). Dall’analisi post-mortem di tessuti cerebrali di pazienti affetti da Alzheimer (solo in tale momento si può confermare la diagnosi clinica da un punto di vista anatomo-patologico), si è potuto riscontrare un accumulo extracellulare di una proteina, chiamata Beta-amiloide. La APP (Amyloid precursor protein, Proteina Progenitrice dell’Amiloide) che viene prodotta è degradata durante il processo di trasporto sulla superficie cellulare (processo di degradazione della APP) e vede coinvolti tre enzimi che operano tagli proteolitici: la α-secretasi e la β-secretasi in un primo momento e successivamente la γ-secretasi. Attraverso due tagli successivi operati prima dall’α-secretasi e poi dall’γ-secretasi, viene prodotto un peptide innocuo chiamato p3. La β-secretasi opera un taglio differente che, in seguito al successivo taglio da parte della γ-secretasi, porta alla produzione (pathway amiloidogenico) di due peptidi di 40 e 42 amminoacidi, chiamati beta-amiloide (Aβ40 e Aβ42): il secondo (Aβ42) è considerato il più tossico a livello neuronale. Nei soggetti sani il processo di degradazione della APP sembra essere operato principalmente dalla α-secretasi. Per motivi non totalmente chiariti, nei soggetti malati l’enzima che interviene sull’APP non è l’α-secretasi ma la β-secretasi, con una larga produzione di proteina beta-amiloide. Tale β-amiloide non presenta le caratteristiche biologiche della forma naturale, ma ha addirittura un effetto tossico sul neurone; ciò è già di per sé un aspetto atipico per una patologia amiloide, nelle quali generalmente il danno è mediato da aspetti citolesivi, compressivi e trofici dati dal deposito fibrillare stesso (il frammento amiloide è generalmente inerte da un punto di vista funzionale fisiopatologico). Alla morte del neurone (dovuta, nelle prime fasi, all’effetto tossico sopracitato) i frammenti amiloidi vengono liberati nello spazio extracellulare tendendo a depositarsi in aggregati fibrillari insolubili via via sempre più grandi, andando a formare le cosiddette placche amiloidi, rilevabili all’esame istologico. Tali placche neuronali innescano un processo reattivo infiammatorio mediato da astrociti e microglia, attivando una risposta immunitaria richiamando macrofagi e neutrofili, i quali produrranno citochine, interleuchine e TNF-α che danneggiano irreversibilmente i neuroni.
Leggi anche:
- Malattia di Alzheimer: screening e diagnosi nelle fasi iniziali della malattia
- Intervento psicosociale e cognitivo nel paziente con malattia di Alzheimer
Proteina Tau
Ulteriori studi mettono in evidenza che nei malati di Alzheimer interviene un ulteriore meccanismo patologico: all’interno dei neuroni una Proteina Tau, fosforilata in maniera anomala, si accumula nei cosiddetti “aggregati neurofibrillari” (o ammassi neurofibrillari). Particolarmente colpiti da questo processo patologico sono i neuroni colinergici, specialmente quelli delle aree corticali, sottocorticali e, tra queste ultime, le aree ippocampali. In particolare, l’ippocampo è una struttura encefalica che svolge un ruolo fondamentale nell’apprendimento e nei processi di memorizzazione; perciò la distruzione dei neuroni di queste zone è ritenuta essere la causa principale della perdita di memoria dei malati. Ipotesi più recenti sulle cause emergono da alcuni studi pubblicati nel 2014, dove si evidenzia l’associazione dell’insorgenza precoce della malattia con la presenza di rame non ceruloplasminico nel sangue.
Leggi anche:
- Malattia di Alzheimer: cura farmacologica
- Sistema nervoso: com’è fatto, a che serve e come funziona
- Differenza tra malattiarbo di Alzheimer, demenza senile, vascolare e reversibile
- Differenza tra malattia di Alzheimer e malattia di Parkinson: sintomi comuni e diversi
- Cervelletto: anatomia esterna ed interna
- Cervelletto: le lesioni cerebellari più comuni
- Le funzioni del cervelletto: apprendimento e correzione dei movimenti del corpo
Dott. Emilio Alessio Loiacono
Medico Chirurgo
Direttore dello Staff di Medicina OnLine
