
 Con “sindrome di Pierre Robin” (o “sequenza di Pierre Robin”) si indica un gruppo di anormalità congenite che può presentarsi sia come sindrome distinta sia come parte di un’altra patologia o sindrome latente, come ad esempio comunemente la sindrome di Stickler o anche la sindrome velocardiofacciale o la sindrome alcolica fetale o la sindrome di Treacher Collins.
Con “sindrome di Pierre Robin” (o “sequenza di Pierre Robin”) si indica un gruppo di anormalità congenite che può presentarsi sia come sindrome distinta sia come parte di un’altra patologia o sindrome latente, come ad esempio comunemente la sindrome di Stickler o anche la sindrome velocardiofacciale o la sindrome alcolica fetale o la sindrome di Treacher Collins.
Frequenza
La sindrome di Pierre Robin ha un’incidenza variabile da 1 su 8500 a 1 su 30.000.
Cause
La diagnosi della sindrome è complessa, dato che alcuni di questi casi rientrano in una sindrome più complessa nosologicamente diversa. La sequenza di Pierre Robin isolata (in assenza di altre malformazioni associate) è presente in circa la metà dei casi. Circa il 10% delle forme isolate è familiare, ma il gene(i)-malattia non è ancora noto. Ancora non è noto come questa anormalità possa verificarsi nei neonati; una teoria sostiene che, ad un certo momento, durante la fase di formazione delle ossa del feto, la punta della mandibola si attacca nel punto dove le clavicole si uniscono (sterno), impedendo così la crescita delle ossa mascellari. Si pensa che, a circa 12-14 settimane di gestazione, quando il feto inizia a muoversi, lo spostarsi della testa fa sì che la mandibola sporga dalle clavicole. Da quel momento in poi, la mandibola del feto cresce come normalmente, con il risultato che, alla nascita, la mandibola del bambino è più piccola di quanto sarebbe stata con uno sviluppo normale, sebbene continui a svilupparsi con un tasso normale fino a che il bambino termina lo sviluppo.
Sintomi e segni
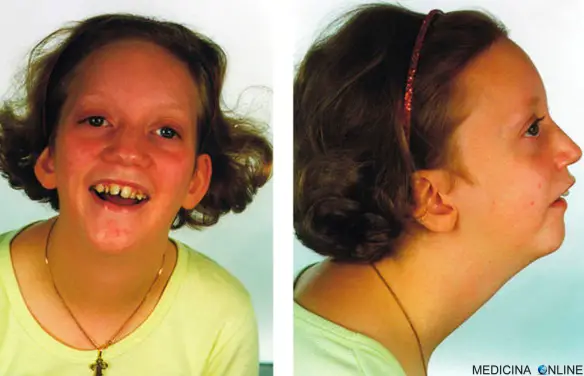
La sindrome di Pierre Robin si caratterizza per una mandibola insolitamente piccola (micrognazia), (glossoptosi), e ostruzione delle vie aeree superiori. La chiusura incompleta del palato (palatoschisi), è presente nella maggioranza dei pazienti, ed esso è di solito a forma di U.
La sindrome di Pierre Robin è caratterizzata da una triade di anomalie della morfologia oro-facciale:
- retrognazia e micrognazia (mandibola piccola e retratta);
- glossoptosi (disposizione posteriore o retrazione della lingua):
- palatoschisi (schisi della parte mediale posteriore del palato molle).
L’ipoplasia mandibolare nelle prime settimane di vita fetale obbliga la lingua a una posizione sollevata nel cavo orale, che ostacola la fusione delle lamine palatine. Le cause di questa anomalia dello sviluppo mandibolare sono diverse, ma raramente è implicato primitivamente un difetto osseo. Nella maggior parte dei casi la malformazione della mandibola è secondaria alla ipomobilità oro-facciale fetale, generalmente correlata a un difetto funzionale del romboencefalo (cervello posteriore). Questo spiega la frequenza e la gravità del quadro clinico nei neonati, che comporta problemi di suzione, deglutizione e respirazione, con difficoltà nell’allattamento, anomala deglutizione, alterazioni della motilità esofagea, ostruzione respiratoria glossofaringo-laringea e sincope vagale.
Diagnosi
La patologia generalmente è diagnosticata poco dopo la nascita, su base clinica.
Leggi anche:
- Sindrome di Möbius: cause, sintomi, diagnosi e terapia
- Sindrome di Stickler: cause, sintomi, diagnosi, cure, prognosi
- Sindrome di Pfeiffer: cause, sintomi, diagnosi, cure, prognosi
Terapia
I problemi medici più importanti sono la difficoltà alla respirazione e alla nutrizione. I neonati affetti necessitano molto spesso di assistenza per la nutrizione, ad esempio necessitando di rimanere in posizione laterale, necessitando ciucciotti o cucchiai speciali, e spesso necessitano di nutrizione con un sondino naso-gastrico o di un’integrazione alimentare adeguata dovuta alla lenta alimentazione. Ciò è causato dalla difficoltà di formare i l vuoto nella cavità orale a causa del palato diviso, oltre alla respirazione difficoltosa dovuta allo spostamento indietro della lingua. Gli infanti, se colpiti moderatamente o severamente, possono occasionalmente necessitare una cannula naso-faringea o, più raramente, intubazione endotracheale o tracheostomia per risolvere l’ostruzione delle vie aeree.
Il palato diviso in genere è curato tra l’età di 6 mesi e ½ e 2 anni con un intervento di chirurgia plastica o maxillofacciale. In molti centri adesso c’è un’équipe per il labbro leporino e per la palatoschisi che comprende entrambe le specialità, come anche un coordinatore, un terapista del linguaggio, un ortodontista, talvolta uno psicologo, un audiologo, un chirurgo otorinolaringoiatra e personale infermieristico. La glossoptosi e micrognazia generalmente non richiedono la chirurgia; in alcuni casi la dis-trazione della mandibola è necessaria per aiutare respirazione e nutrizione.
Prognosi
Se il trattamento del quadro clinico è efficace nel primo anno di vita, la prognosi è buona. I bambini affetti da sindrome di Pierre Robin di solito raggiungono pieno sviluppo e statura. Comunque, è stato riscontrato che il bambino è spesso leggermente di taglia sotto la media, a causa di sviluppo incompleto dovuto a ipossia cronica relativa a ostruzione delle vie aeree superiori come pure alla scarsa nutrizione dovute a difficoltà di alimentazione.
Leggi anche:
- Le malattie genetiche più diffuse al mondo
- Differenza tra autosomica dominante e recessiva con esempi
- Quando una mammella non si sviluppa: la Sindrome di Poland
- Sindrome di Down: cause, sintomi in gravidanza e nei neonati
- Anemia falciforme: cosa significa, cause, sintomi e cure
- Sindrome di Turner: cariotipo, cause, sintomi e segni caratteristici
- Sindrome di Klinefelter: cariotipo, cause, sintomi e cura
- Sindrome di Noonan: cause, sintomi nel neonato, aspettative di vita
- Sindrome di Bloom: cause, sintomi, diagnosi e terapia
- Fibrosi cistica polmonare: cos’è, sintomi in neonati e bambini, cure
- Malattia di Huntington: cos’è, ereditarietà, come si trasmette, età di insorgenza
- Aracnodattilia, segno del pollice e del polso, Sindrome di Marfan e di Beals
- Ectrodattilia: cause, cure ed immagini
- Polidattilia; cause, ereditarietà, sindromica e chirurgia
- Sindrome di Beals e padiglione auricolare “accartocciato”: sintomi e cure
- Differenza tra polidattilia, sindattilia, ectrodattilia, oligodattilia e aracnodattilia
- Cos’è un cromosoma ed a che serve?
- Sindrome dell’idiota sapiente: cause, caratteristiche e sintomi
- Sindrome del tramonto o del crepuscolo: cause, sintomi e cura
- Ritardo mentale nei bambini lieve, moderato, grave: si guarisce?
- Che cos’è l’intelligenza umana: definizione, significato e psicologia
- Quoziente d’intelligenza: valori, significato, test ed ereditarietà
- Problem solving: cos’è, caratteristiche, tecniche, fasi ed esempi
- Sindrome di Tourette: cause, sintomi, diagnosi e trattamento
- Sindrome di Tourette: si può guarire definitivamente? Come si guarisce?
- Differenza tra allele dominante e recessivo
- Differenza tra omozigote ed eterozigote
- Differenza tra gene e allele
- Differenza tra genotipo e fenotipo
Lo Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o unisciti al nostro gruppo Facebook o ancora seguici su Twitter, su Instagram o su Pinterest, grazie!
