
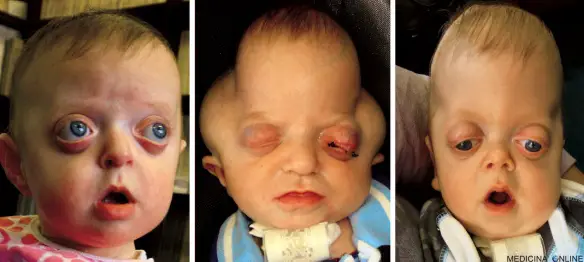
La sindrome di Pfeiffer di tipo 1 (a sinistra), di tipo 2 (al centro) e di tipo 3 (a destra).
La sindrome di Pfeiffer (o “acrocefalosindattilia tipo 5”) è una malattia genetica a trasmissione autosomica dominante molto rara caratterizzata dalla fusione prematura di alcune ossa delcranio (con interessamento della testa e del viso) con malformazioni alle mani e ai piedi di gravità variabile: è una delle forme più frequenti di acrocefalosindattilia. La sindrome di Pfeiffer è una craniostenosi associata a una mutazione del gene FGPR; comprende diversi tipi a seconda del tipo di gene coinvolto. Le suture del cranio che si fondono in questa sindrome sono quelle coronali e a volte sagittali.
Frequenza
La sindrome di Pfeiffer è molto rara: colpisce 1 su 100.000 nati.
Cause
La sindrome di Pfeiffer è fortemente associata a mutazioni del recettore 1 del fattore di crescita dei fibroblasti (FGFR1) sul cromosoma 8 o del recettore 2 del fattore di crescita dei fibroblasti (FGFR2) sul cromosoma 10. Questi geni codificano per i recettori del fattore di crescita dei fibroblasti, che sono importanti per il normale sviluppo osseo. Si ritiene che l’età paterna avanzata sia un fattore di rischio per alcuni casi di sindrome di Pfeiffer a causa di un aumento delle mutazioni nel liquido spermatico man mano che gli uomini invecchiano.
Leggi anche:
- Sindrome di Möbius: cause, sintomi, diagnosi e terapia
- Sindrome di Pierre Robin: cause, sintomi, diagnosi, cure, prognosi
- Sindrome di Stickler: cause, sintomi, diagnosi, cure, prognosi
Segni e sintomi
Molti dei tratti caratteristici del viso derivano dalla fusione prematura delle ossa del cranio (craniostenosi). La testa non è in grado di crescere correttamente, il che porta a una fronte alta prominente (brachicefalia) e a protusione (proptosi) e distanziamento tra i bulbi oculari (ipertelorismo). Inoltre, vi è una mascella superiore sottosviluppata (ipoplasia del mascellare). Circa il 50% dei bambini con sindrome di Pfeiffer ha perdita dell’udito, e sono comuni anche problemi dentali. Nei soggetti con sindrome di Pfeiffer sono presente anomalie alle mani e ai piedi, in particolare alle dita. Sono ad esempio comuni dita insolitamente corte (brachidattilia) oppure fuse tra di loro (sindattilia). Ognuno dei tre tipi ha delle caratteristiche particolari, che verranno illustrate nel prossimo paragrafo.
Classificazione
La classificazione clinica più ampiamente accettata della sindrome di Pfeiffer è stata pubblicata da M. Michael Cohen nel 1993. Cohen ha suddiviso la sindrome in tre tipi, possibilmente sovrapposti:
- sindrome di Pfeiffer di tipo 1 (sindrome di Pfeiffer classica): include craniostenosi e un’alterazione della crescita scheletrica della faccia mediale. Questo tipo è ereditato secondo un modello autosomico dominante. La maggior parte delle persone con sindrome di Pfeiffer di tipo 1 ha un’intelligenza normale, durata di vita normale, bassa portata della ipoplasia medio-facciale, brachicefalia, anomalie di basso grado di mani e dei piedi;
- sindrome di Pfeiffer di tipo 2: presenta un cranio a forma di quadrifoglio (Cloverleaf skull o Kleeblattschädel) a causa di un’estensiva fusione delle ossa craniche e di una grave proptosi. Questo tipo si presenta sporadicamente (cioè non sembra essere ereditato), ha una prognosi infausta e una grave compromissione neurologica, generalmente con morte precoce;
- sindrome di Pfeiffer di tipo 3: include craniostenosi e grave proptosi. Anche questo tipo si presenta sporadicamente (cioè non sembra essere ereditato), ha una prognosi infausta e una grave compromissione neurologica, generalmente con morte precoce.
Trattamento e prognosi
Il principale problema è la fusione precoce del cranio, che può essere corretta con una serie di procedure chirurgiche, spesso entro i primi tre mesi dalla nascita. Gli interventi chirurgici successivi sono necessari per correggere le deformità respiratorie e facciali. I bambini con sindrome di Pfeiffer di tipo 2 e 3 hanno un rischio maggiore di sviluppare disturbi neurologici e una ridotta aspettativa di vita” rispetto ai bambini con sindrome di Pfeiffer di tipo 1, ma se trattati, possono verificarsi esiti favorevoli. Nei casi più gravi, le complicanze respiratorie e neurologiche portano a morte prematura.
Leggi anche:
- Le malattie genetiche più diffuse al mondo
- Differenza tra autosomica dominante e recessiva con esempi
- Quando una mammella non si sviluppa: la Sindrome di Poland
- Sindrome di Down: cause, sintomi in gravidanza e nei neonati
- Anemia falciforme: cosa significa, cause, sintomi e cure
- Sindrome di Turner: cariotipo, cause, sintomi e segni caratteristici
- Sindrome di Klinefelter: cariotipo, cause, sintomi e cura
- Sindrome di Noonan: cause, sintomi nel neonato, aspettative di vita
- Sindrome di Bloom: cause, sintomi, diagnosi e terapia
- Fibrosi cistica polmonare: cos’è, sintomi in neonati e bambini, cure
- Malattia di Huntington: cos’è, ereditarietà, come si trasmette, età di insorgenza
- Aracnodattilia, segno del pollice e del polso, Sindrome di Marfan e di Beals
- Ectrodattilia: cause, cure ed immagini
- Polidattilia; cause, ereditarietà, sindromica e chirurgia
- Sindrome di Beals e padiglione auricolare “accartocciato”: sintomi e cure
- Differenza tra polidattilia, sindattilia, ectrodattilia, oligodattilia e aracnodattilia
- Cos’è un cromosoma ed a che serve?
- Sindrome dell’idiota sapiente: cause, caratteristiche e sintomi
- Sindrome del tramonto o del crepuscolo: cause, sintomi e cura
- Ritardo mentale nei bambini lieve, moderato, grave: si guarisce?
- Che cos’è l’intelligenza umana: definizione, significato e psicologia
- Quoziente d’intelligenza: valori, significato, test ed ereditarietà
- Problem solving: cos’è, caratteristiche, tecniche, fasi ed esempi
- Sindrome di Tourette: cause, sintomi, diagnosi e trattamento
- Sindrome di Tourette: si può guarire definitivamente? Come si guarisce?
- Differenza tra allele dominante e recessivo
- Differenza tra omozigote ed eterozigote
- Differenza tra gene e allele
- Differenza tra genotipo e fenotipo
Lo Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o unisciti al nostro gruppo Facebook o ancora seguici su Twitter, su Instagram o su Pinterest, grazie!
