
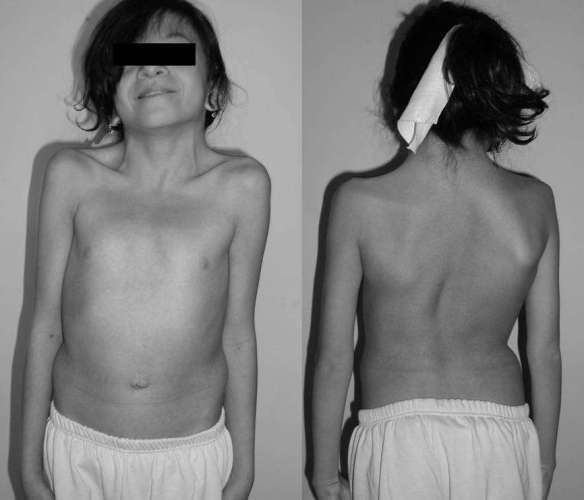
 La sindrome di Noonan è una malattia genetica dalle caratteristiche cliniche particolarmente variabili, con un’incidenza stimata di un caso ogni 1000-2500 nati. Le manifestazioni più frequenti includono cardiopatie congenite (nel 70% dei pazienti), bassa statura, un aspetto caratteristico del volto (abbassamento delle palpebre, occhi distanziati, orecchie grandi e ruotate posteriormente), malformazioni del torace, deficit cognitivi variabili, tendenza al sanguinamento e criptorchidismo (mancata discesa dei testicoli nello scroto).
La sindrome di Noonan è una malattia genetica dalle caratteristiche cliniche particolarmente variabili, con un’incidenza stimata di un caso ogni 1000-2500 nati. Le manifestazioni più frequenti includono cardiopatie congenite (nel 70% dei pazienti), bassa statura, un aspetto caratteristico del volto (abbassamento delle palpebre, occhi distanziati, orecchie grandi e ruotate posteriormente), malformazioni del torace, deficit cognitivi variabili, tendenza al sanguinamento e criptorchidismo (mancata discesa dei testicoli nello scroto).
Come si trasmette la sindrome di Noonan?
La sindrome può essere sporadica (il difetto genetico responsabile insorge in modo spontaneo) oppure ereditata dai genitori. In questo caso la trasmissione avviene con modalità autosomica dominante: è sufficiente cioè una copia alterata del gene per sviluppare la malattia. In circa il 50% dei casi, la sindrome è causata da mutazioni del gene PTPN11, mentre in una piccola percentuale di pazienti sono state identificate mutazioni nei geni KRAS, NRAS, RAF1, BRAF e SOS1. Nel complesso, le mutazioni nei geni identificati si riscontrano in circa il 75% dei pazienti con diagnosi clinica della malattia. Mutazioni nei geni SHOC2 e CBL sono state riscontrate in soggetti con caratteristiche cliniche parzialmente sovrapponibili alla sindrome di Noonan.
Leggi anche:
- Le malattie genetiche più diffuse al mondo
- Sindrome di Turner: cariotipo, cause, sintomi e segni caratteristici
- Sindrome di Klinefelter: cariotipo, cause, sintomi e cura
- Sindrome di Down: cause, sintomi in gravidanza e nei neonati
- Fibrosi cistica polmonare: cos’è, sintomi in neonati e bambini, cure
- Malattia di Huntington: cos’è, ereditarietà, come si trasmette, età di insorgenza
- Anemia falciforme: cosa significa, cause, sintomi e cure
- Differenze tra la distrofia muscolare di Duchenne e di Becker
- Talassemia: cos’è, sintomi, cure, differenti tipi ed alimentazione
- Celiachia: cos’è il glutine, in quali alimenti è contenuto ed in quali no?
- Sindrome di Bloom: cause, sintomi, diagnosi e terapia
Come avviene la diagnosi della sindrome di Noonan?
La diagnosi avviene in base alle caratteristiche cliniche ed eventualmente all’analisi della storia familiare e può essere confermata con un’indagine genetica (ricerca di mutazioni nei geni PTPN11, RAF1, KRAS, SOS1, NRAS e BRAF). Va comunque ricordato che, per molti pazienti, la causa genetica della malattia non è ancora stata identificata. Inoltre, vista la variabilità clinica della malattia, l’analisi molecolare di altri geni implicati in malattie correlate (HRAS, MEK1, MEK2, CBL e SHOC2) può indirizzare verso una più corretta valutazione clinica. Se una mutazione responsabile è già stata identificata in un componente della famiglia, si può effettuare una diagnosi prenatale con l’indagine genetica del feto attraverso villocentesi o amniocentesi. L’età media alla diagnosi è di nove anni.
Quali sono le possibilità di cura attualmente disponibili per la sindrome di Noonan?
Non esiste una terapia specifica, anche se studi preclinici su modelli animali sono in corso per valutare l’efficacia di alcuni farmaci che inibiscono in maniera specifica l’attività degli enzimi “mutati” responsabili della malattia. Alcune lesioni cardiache devono essere corrette per via chirurgica, mentre in alcuni casi è consigliata l’assunzione di ormone della crescita. In generale, i bambini affetti dalla sindrome vanno tenuti sotto controllo per quanto riguarda la funzionalità cardiaca, la crescita, lo sviluppo motorio, la coagulazione del sangue. Se seguita in modo adeguato, la maggior parte dei bambini colpiti cresce normalmente e raggiunge senza problemi l’età adulta.
Aspettative di vita
La speranza di vita è generalmente normale (paragonabile al resto della popolazione) se sono assenti difetti cardiaci congeniti importanti. L’aspettativa di vita diminuisce all’aumentare della gravità delle patologie cardiache.
Leggi anche:
- Cos’è un cromosoma ed a che serve?
- Sindrome dell’idiota sapiente: cause, caratteristiche e sintomi
- Sindrome del tramonto o del crepuscolo: cause, sintomi e cura
- Ritardo mentale nei bambini lieve, moderato, grave: si guarisce?
- Che cos’è l’intelligenza umana: definizione, significato e psicologia
- Quoziente d’intelligenza: valori, significato, test ed ereditarietà
- Problem solving: cos’è, caratteristiche, tecniche, fasi ed esempi
- Sindrome di Tourette: cause, sintomi, diagnosi e trattamento
- Sindrome di Tourette: si può guarire definitivamente? Come si guarisce?
- Differenza tra allele dominante e recessivo
- Differenza tra omozigote ed eterozigote
- Differenza tra gene e allele
- Differenza tra genotipo e fenotipo
- Quanti cromosomi hanno esseri umani, scimmie, cani, gatti e topi?
- Quanti cromosomi ha chi è affetto da Sindrome di Down?
- Cos’è un gene ed a che serve?
- Cosa sono gli alleli ed a che servono?
- Differenza tra cellule eucariote e procariote
- Virus e virioni: cosa sono, come sono fatti, come funzionano e come si riproducono
- Differenza tra cellula aploide e diploide con esempi
- Riproduzione cellulare e ciclo cellulare
- Meiosi: spiegazione di tutte tappe
- Mitosi: spiegazione delle quattro fasi
- Differenza tra mitocondri e cloroplasti
- Differenza tra citosol e citoplasma
- Differenza tra virus e batteri: chi è più pericoloso? Diagnosi, sintomi e terapia
- Organelli (organuli) citoplasmatici della cellula animale: cosa sono ed a che servono?
- Mitocondri: definizione, dimensioni e funzioni
- Citoscheletro: funzioni e struttura
- Ribosomi e reticolo endoplasmatico: cosa sono e che funzioni svolgono?
- Nucleo cellulare: funzioni, dimensioni e membrane nucleari
- Lisosomi: cosa sono? Significato e dimensioni
- Perossisomi: definizione e funzioni
- Membrana plasmatica: definizione e funzioni
- Apparato del Golgi: spiegazione semplice e funzioni
- Citosol: definizione e funzioni
Lo staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o seguici su Twitter, su Instagram o su Pinterest, grazie!
