
 La “sindrome di Behçet“, anche denominata “malattia della Via della Seta” o “malattia di Behçet” o “morbo di Behçet” (in inglese “Behçet disease” o “Behçet’s syndrome” o “morbus Behçet” o “Silk Road disease“) è una rarissima sindrome infiammatoria multisistemica cronica e recidivante caratterizzata da afte orali, genitali, uveite, tromboflebite e che frequentemente coinvolge le articolazioni, la cute, il sistema nervoso centrale e il tratto gastrointestinale. Le lesioni sulle mucose possono durare da pochi giorni fino a una settimana o più. Meno comunemente ci possono essere infiammazioni del cervello o del midollo spinale, coaguli di sangue, aneurismi o cecità. La sindrome alterna periodi sintomatici a periodi di remissione dei sintomi.
La “sindrome di Behçet“, anche denominata “malattia della Via della Seta” o “malattia di Behçet” o “morbo di Behçet” (in inglese “Behçet disease” o “Behçet’s syndrome” o “morbus Behçet” o “Silk Road disease“) è una rarissima sindrome infiammatoria multisistemica cronica e recidivante caratterizzata da afte orali, genitali, uveite, tromboflebite e che frequentemente coinvolge le articolazioni, la cute, il sistema nervoso centrale e il tratto gastrointestinale. Le lesioni sulle mucose possono durare da pochi giorni fino a una settimana o più. Meno comunemente ci possono essere infiammazioni del cervello o del midollo spinale, coaguli di sangue, aneurismi o cecità. La sindrome alterna periodi sintomatici a periodi di remissione dei sintomi.
Eponimo
La sindrome di Behçet deve il suo nome al dermatologo turco Hulusi Behçet (1889-1948), che per primo riconobbe i tre sintomi principali della sindrome in uno dei suoi pazienti nel 1924 e riportò le sue ricerche sulla malattia nel Journal of Skin and Venereal Diseases in 1936.
Storia
I sintomi della sindrome di Behçet potrebbero essere stati descritti da Ippocrate nel V secolo a.C., nella sua Epidemion (libro 3, caso 7). La prima moderna descrizione formale dei sintomi della sindrome di Behçet fu fatta da H. Planner e F. Remenovsky e pubblicata nel 1922 nell’Archiv für Dermatologie und Syphilis. Dopo che Hulusi Behçet riconobbe i tre sintomi principali della sindrome, il nome “morbo di Behçet” fu formalmente adottato al Congresso Internazionale di Dermatologia a Ginevra nel settembre 1947. Alcune fonti usano il termine “sindrome di Adamantiades” o “sindrome di Adamantiades-Behçet“, per il lavoro svolto dal ricercatore Benediktos Adamantiades, tuttavia l’attuale denominazione standard è “malattia di Behçet”. Nel 1991, alcuni ricercatoridell’Arabia Saudita hanno descritto la malattia neuro-Behçet (anche chiamata “sindrome neuro-Behçet“), un coinvolgimento neurologico nella malattia di Behçet, considerata una delle manifestazioni più devastanti della malattia.
Epidemiologia
La sindrome di Behçet colpisce prevalentemente tra i 20 e i 40 anni. A livello globale, i maschi sono colpiti più frequentemente delle femmine. Si stima che a circa 15.000-20.000 americani sia stata diagnosticata questa malattia. Nel Regno Unito, si stima che ci sia circa 1 caso ogni 100.000 persone. In Italia l’incidenza è di 2,4 casi ogni milione di abitanti e la prevalenza di 3,8/100 000. Sebbene molto raro negli Stati Uniti e in Europa, è più comune in Medio Oriente e in Asia. In Turchia, ad esempio, ne sono colpiti circa 2 su 1.000.
Cause e fattori di rischio
Le cause della sindrome di Behçet sono attualmente sconosciute anche se si pensa che l’eziologia sia relativa a cause autoimmunitarie e genetiche. La sindrome di Behçet non è contagiosa. Il coinvolgimento di un sottogruppo di cellule T (Th17) sembra essere importante nella patogenesi. Alla base del meccanismo autoimmunitario sembra ci sia una componente genetica, poiché i parenti di primo grado dei pazienti affetti sono spesso colpiti in misura superiore a quella prevista per la popolazione generale. La ricerca suggerisce che precedenti infezioni potrebbero provocare le risposte autoimmuni presenti nella malattia di Behçet. Le proteine da shock termico (HSP) sono presenti in alcuni batteri e fungono da “segnale di pericolo” per il sistema immunitario. Tuttavia, alcuni HSP condividono una somiglianza nei batteri e negli esseri umani. Gli anticorpi anti-HSP60 e anti-HSP65 che prendono di mira le HSP prodotte da Streptococchi (inclusi S. sanguinis e S. pyogenes) e Mycobacterium tuberculosis possono colpire anche le HSP umane, portando a risposte immunitarie legate all’uveite e a vari sintomi mostrati nella malattia neurologica di Behçet (“malattia neuro-Behçet” o “sindrome neuro-Behçet“). È stata segnalata un’associazione con la famiglia di geni GIMAP (“GTPasi della proteina associata all’immunità”) sul braccio lungo del cromosoma 7 (7q36.1). I geni implicati erano GIMAP1, GIMAP2 e GIMAP4.
Fisiopatologia
La malattia di Behçet è considerata più diffusa nelle aree circostanti le antiche rotte commerciali della seta in Medio Oriente e in Asia centrale., tanto che viene anche chiamata “malattia della Via della Seta”, tuttavia questa malattia non è limitata alle persone di queste regioni. Un gran numero di studi sierologici mostra un legame tra la malattia e l’HLA-B51. L’HLA-B51 è più frequente dal Medio Oriente alla Siberia sudorientale, ma l’incidenza di B51 in alcuni studi era 3 volte superiore rispetto alla popolazione normale. Tuttavia, B51 tende a non essere trovato nella malattia quando è coinvolta una variante del gene SUMO4 ed i sintomi sembrano essere più lievi quando è presente HLA-B27. Al momento, non è stata ancora confermata un’origine infettiva simile che porti alla malattia di Behçet, ma è stato riscontrato che alcuni ceppi di S. sanguinis hanno un’antigenicità omologa. La vasculite con conseguente occlusione dei vasi che irrorano il nervo ottico può essere la causa della neuropatia ottica acuta e dell’atrofia ottica progressiva nella malattia di Behçet. La valutazione istologica in un caso segnalato di neuropatia ottica acuta ha dimostrato la sostituzione della porzione assonale del nervo ottico con astrociti fibrosi senza alterazioni retiniche. Il coinvolgimento del sistema nervoso centrale nella malattia di Behçet può portare a ipertensione endocranica più comunemente a causa di trombosi del seno venoso durale e successiva atrofia ottica secondaria.
Sintomi, segni e complicanze
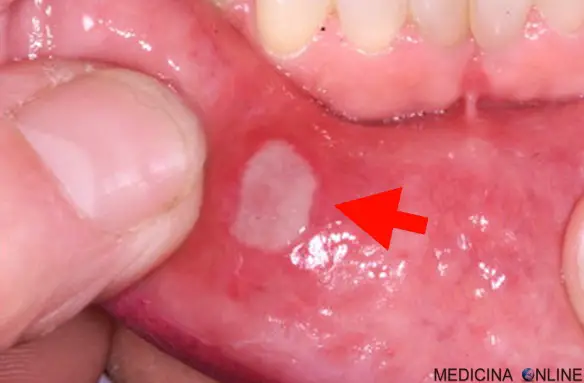
L’esordio dei sintomi avviene di solito tra i 20 ei 40 anni. Quasi tutte le persone con la malattia di Behçet presentano qualche forma di ulcerazione dolorosa all’interno della bocca. Le lesioni orali sono simili a quelle riscontrate nella malattia infiammatoria intestinale e possono essere recidivanti. Frequenti anche le ulcerazioni genitali, generalmente dolorose, che di solito si sviluppano attorno all’ano, alla vulva o allo scroto e causano cicatrici nel 75% dei pazienti. Inoltre, i pazienti possono presentare eritema nodoso, vasculite pustolosa cutanea e lesioni simili al pioderma gangrenoso.
La sindrome porta anche a sintomi e segni relativi all’apparato visivo: la malattia infiammatoria dell’occhio può svilupparsi all’inizio del decorso della malattia e portare alla perdita permanente della vista nel 20% dei casi. Il coinvolgimento oculare può essere sotto forma di uveite posteriore, uveite anteriore o vasculite retinica. L’uveite anteriore si presenta con occhi dolorosi, arrossamento congiuntivale, ipopione e ridotta acuità visiva, mentre l’uveite posteriore si presenta con ridotta acuità visiva indolore e corpi mobili del campo visivo. Una rara forma di coinvolgimento oculare in questa sindrome è la vasculite retinica che si presenta con diminuzione indolore della vista con la possibilità di corpi mobili o difetti del campo visivo. Il coinvolgimento del nervo ottico nella malattia di Behçet è raro e si presenta tipicamente come atrofia ottica progressiva e perdita della vista. Tuttavia, sono stati segnalati anche casi di neuropatia ottica acuta (in particolare neuropatia ottica ischemica anteriore). L’atrofia del nervo ottico è stata identificata come la causa più comune di disabilità visiva permanente. Il papilledema come conseguenza della trombosi del seno durale e l’atrofia derivante dalla malattia della retina, sono stati caratterizzati come cause secondarie dell’atrofia del nervo ottico nella malattia di Behçet. Segni e sintomi di neuropatia ottica acuta includono:
- perdita indolore della vista che può interessare uno o entrambi gli occhi,
- ridotta acuità visiva,
- ridotta visione dei colori,
- difetto pupillare afferente relativo,
- scotoma centrale,
- disco ottico gonfio,
- edema maculare,
- dolore retrobulbare.
Quando questi sintomi si verificano con concomitanti ulcerazioni mucocutanee, sollevano il sospetto di neuropatia ottica acuta nella malattia di Behçet. L’atrofia ottica progressiva può causare una riduzione dell’acuità visiva o della visione dei colori. Può essere presente ipertensione endocranica con papilledema. Può verificarsi episclerite, che provoca arrossamento degli occhi e lieve dolore, senza un impatto significativo sulla vista.
Le manifestazioni gastrointestinali includono dolore addominale, nausea e diarrea con o senza sangue e spesso coinvolgono la valvola ileocecale. Alcuni pazienti sperimentano dolorabilità addominale, gonfiore e disagio addominale generale. Quando la malattia è lieve, può somigliare clinicamente alla sindrome dell’intestino irritabile; i casi più gravi presentano somiglianze con le malattie infiammatorie intestinali come la colite ulcerosa o il morbo di Crohn.
Il coinvolgimento polmonare è tipicamente sotto forma di emottisi, pleurite, tosse o febbre, e nei casi più gravi può essere pericoloso per la vita se l’arteria polmonare di uscita sviluppa un aneurisma che si rompe causando un grave collasso vascolare e la morte per sanguinamento nei polmoni. Noduli, consolidamenti, cavità e lesioni a vetro smerigliato sono comuni nei pazienti con interessamento polmonare. Può verificarsi trombosi dell’arteria polmonare.
Possibile anche il coinvolgimento delle articolazioni. L’artrite è presente in più della metà delle persone e di solito è una poli o oligoartrite non erosiva principalmente delle grandi articolazioni degli arti inferiori.
Il coinvolgimento del sistema nervoso centrale (SNC) si verifica più spesso come meningoencefalite cronica. Le lesioni tendono a verificarsi nel tronco encefalico, nei gangli della base e nella sostanza bianca emisferica profonda e possono assomigliare a quelle della sclerosi multipla (SM). L’atrofia del tronco cerebrale è osservata nei casi cronici. Il quadro neurologico prende il nome di “malattia neurogica di Behçet” o “malattia neuro-Behçet” o “sindrome neuro-Behçet” (in inglese “neuro-Behçet’s disease“). I coinvolgimenti neurologici vanno dalla meningite asettica alla trombosi vascolare come la trombosi del seno durale e la sindrome cerebrale organica che si manifesta con confusione, convulsioni e perdita di memoria. Ad essa è spesso associata un’improvvisa perdita dell’udito (sensorineurale).Spesso compaiono in ritardo nella progressione della malattia ma sono associati a una prognosi infausta.
Possibile anche il coinvolgimento del cuore e dei vasi sanguigni. La pericardite è una manifestazione cardiaca frequente. Si può anche osservare rigurgito aortico cronico dovuto a malattia della radice aortica. Sebbene raro, è stato segnalato un caso di infarto del miocardio con trombosi coronarica acuta identificata angiograficamente. I problemi dei vasi sanguigni si osservano nel 7-29% delle persone con lesioni arteriose che rappresentano il 15% delle lesioni vascolari. Le lesioni arteriose rappresentano un rischio maggiore. Le lesioni arteriose più comuni sono occlusioni o stenosi e aneurismi o pseudoaneurismi.
La malattia di Behçet può causare infertilità maschile, sia come risultato della condizione stessa, sia come effetto collaterale di farmaci concomitanti come la colchicina, che è nota per abbassare il numero di spermatozoi.
Gravidanza
Nelle pazienti con malattia di Behçet, la gravidanza non ha un effetto negativo sul decorso della malattia di Behçet ed anzi può eventualmente migliorarne il decorso, tuttavia c’è una sostanziale variabilità nel decorso clinico tra pazienti e anche per gravidanze diverse nella stessa paziente. Inoltre la malattia di Behçet conferisce un aumentato rischio di complicanze della gravidanza, di aborto spontaneo e di necessità di taglio cesareo.
Diagnosi
La diagnosi della malattia di Behçet si basa su reperti clinici che includono ulcere orali e genitali, lesioni cutanee come eritema nodoso, acne o follicolite, reperti infiammatori oculari e una reazione di patergia. Secondo le linee guida dell’International Study Group, affinché a un paziente venga diagnosticata la malattia di Behçet, il paziente deve avere ulcere orali (afte) (di qualsiasi forma, dimensione o numero almeno 3 volte in un periodo di 12 mesi) insieme a 2 dei seguenti 4 sintomi e segni:
- infiammazione oculare (irite, uveite, vasculite retinica, cellule del vitreo);
- ulcere genitali (comprese ulcere anali e macchie nella regione genitale e testicoli gonfi o epididimite negli uomini);
- reazione di patergia (papule > 2 mm di diametro 24-48 ore o più dopo la puntura dell’ago). Il test di patergia ha una specificità dal 95% al 100%, ma i risultati sono spesso negativi nei pazienti americani ed europei;
- lesioni cutanee (papulo-pustole, follicolite, eritema nodoso, acne in post-adolescenti non in terapia con corticosteroidi).
Nonostante i criteri inclusivi stabiliti dall’International Study Group, ci sono casi in cui non tutti i criteri possono essere soddisfatti e quindi una diagnosi non può essere raggiunta facilmente. E’ comunque necessario un alto grado di sospetto per un paziente che presenta i seguenti sintomi, segni e condizioni:
- artrite/artralgia;
- problemi cardiovascolari di origine infiammatoria;
- cambiamenti di personalità, psicosi;
- trombosi venosa profonda;
- epididimite;
- stanchezza cronica;
- problemi infiammatori al torace e ai polmoni;
- ulcere della bocca;
- sintomi neurologici;
- problemi di udito o di equilibrio;
- infiammazione dello stomaco o dell’intestino;
- tromboflebite superficiale;
- qualsiasi altro membro della famiglia con una diagnosi di malattia di Behçet.
Sebbene a volte ci si riferisca erroneamente a una diagnosi di esclusione, in alcuni casi la diagnosi può essere raggiunta mediante un esame patologico delle aree colpite. L’acuità visiva o la perdita della visione dei colori con concomitanti lesioni mucocutanee o sintomi sistemici della malattia di Behçet devono nel medico sollevare il sospetto di un coinvolgimento del nervo ottico nella malattia di Behçet e richiedere un esame per la malattia di Behçet se non precedentemente diagnosticato in aggiunta a un esame oculare. I marcatori infiammatori come ESR e CRP possono essere elevati. Un esame oftalmologico completo può includere un esame con lampada a fessura, tomografia a coerenza ottica per rilevare la perdita di nervi, esami del campo visivo, esame fundoscopico per valutare l’atrofia del disco ottico e la malattia della retina, angiografia fundoscopica e potenziali evocati visivi, che possono dimostrare una maggiore latenza. La condizione del nervo ottico può essere identificato sulla risonanza magnetica in alcuni pazienti con neuropatia ottica acuta. Esami neurologici e talvolta psichiatrici possono rendersi necessari. L’analisi del liquido cerebrospinale (“liquor”) può dimostrare un livello proteico elevato con o senza pleiocitosi. L’imaging inclusa l’angiografia può essere indicato per identificare la trombosi del seno venoso durale come causa di ipertensione intracranica e atrofia ottica.
Terapia
Non esiste una cura specifica per la malattia di Behçet: il trattamento attuale ha lo scopo di alleviare i sintomi, ridurre l’infiammazione e controllare il sistema immunitario. I trattamenti possono includere farmaci immunosoppressori come i corticosteroidi e cambiamenti dello stile di vita. Il collutorio alla lidocaina può alleviare il dolore. La colchicina può ridurre la frequenza degli attacchi. La terapia con corticosteroidi ad alte dosi viene spesso utilizzata per le manifestazioni di malattia grave. La terapia anti-TNF come l’infliximab ha mostrato risultati promettenti nel trattamento dell’uveite associata alla malattia. Un altro agente anti-TNF, l’etanercept, può essere utile nelle persone con sintomi principalmente cutanei e delle mucose. L’apremilast può anche essere usato per trattare le ulcere orali associate alla malattia di Behçet. L’interferone alfa-2a può anche essere un trattamento alternativo efficace, in particolare per le ulcere genitali e orali così come per le lesioni oculari. Anche l’azatioprina, se usata in combinazione con l’interferone alfa-2b, si mostra promettente, e la colchicina può essere utile per trattare alcune ulcere genitali, l’eritema nodoso e l’artrite. La benzatina-penicillina può anche ridurre i nuovi attacchi artritici. La talidomide è stata utilizzata anche per il suo effetto immuno-modificante. Dapsone e rebamipide hanno dimostrato, in piccoli studi, di avere risultati benefici per le lesioni mucocutanee. Data la sua rarità, il trattamento ottimale per la neuropatia ottica acuta nella malattia di Behçet non è stato stabilito. L’identificazione e il trattamento precoci sono essenziali. È stata segnalata risposta alla ciclosporina, al triamcinolone perioculare e al metilprednisone EV seguito da prednisone orale, sebbene possano verificarsi ricadute che portano alla perdita irreversibile della vista anche con il trattamento. Gli immunosoppressori come l’interferone-alfa e gli antagonisti del fattore di necrosi tumorale possono migliorare, sebbene non invertire completamente, i sintomi della malattia oculare di Behçet, che può progredire nel tempo nonostante il trattamento. Quando i sintomi sono limitati alla camera anteriore dell’occhio, la prognosi è migliore. Il coinvolgimento posteriore, in particolare il coinvolgimento del nervo ottico, è un indicatore prognostico sfavorevole e può determinare perdita permanente della vista. L’atrofia secondaria del nervo ottico è spesso irreversibile. La puntura lombare o il trattamento chirurgico possono essere necessari per prevenire l’atrofia ottica nei casi di ipertensione endocranica refrattaria al trattamento con immunomodulatori e steroidi. Le IVIG (immunoglobuline per uso endovenoso) potrebbero rappresentare un trattamento per casi gravi o complicati.
Chirurgia
Il trattamento chirurgico delle manifestazioni arteriose della malattia di Behçet comporta molte insidie poiché l’endarterite obliterante dei vasa vasorum provoca l’ispessimento dello strato mediale e la scissione delle fibre di elastina. Pertanto, è probabile che si formino pseudoaneurismi anastomotici, nonché pseudoaneurismi nella sede della puntura in caso di angiografia o trattamento endovascolare; inoltre, può verificarsi un’occlusione precoce dell’innesto. Per questi motivi, il trattamento invasivo non dovrebbe essere eseguito nelle fasi acute e attive della malattia quando l’infiammazione è al suo apice. La valutazione dell’attività della malattia è solitamente basata su sintomi recidivanti, VES (velocità di eritrosedimentazione) e livelli sierici di CRP (proteina C reattiva). Il trattamento endovascolare può essere un’alternativa efficace e più sicura rispetto alla chirurgia a cielo aperto, con minori complicanze postoperatorie, tempi di recupero più rapidi e ridotta necessità di terapia intensiva, offrendo al contempo tassi di pervietà e tassi di successo procedurali paragonabili a quelli della chirurgia. Ciò nonostante, i risultati a lungo termine del trattamento endovascolare nella malattia di Behçet devono ancora essere determinati.
Per approfondire, continua la lettura con: Sindrome neuro-Behçet: cause, sintomi, diagnosi, terapie
Leggi anche:
- Aftosi orale ed afte: cause, cure, rimedi, consigli, cibi consigliati, collutorio
- Uveiti: classificazione, cause, sintomi, diagnosi e terapie
- Tromboflebite superficiale: significato, diagnosi, cure, rischi
- Collutori naturali e fatti in casa per disinfettare la lingua
- Cheilite angolare (boccarola): cause, sintomi, diagnosi, cura, rimedi
- Febbre periodica con stomatite aftosa, faringite e adenite (PFAPA)
- Afte, nevralgia, herpes ed altre cause di dolore alla lingua (glossodinia)
- Lingua bianca e afte nei neonati: cause e cure
- Igiene orale: consigli e prodotti per pulire lingua, denti e bocca
- Come pulire efficacemente la lingua in modo semplice e sicuro
- Come e quando iniziare a lavare i denti ai bambini?
- Quante volte e quando lavare i denti ogni giorno?
- Lavare i denti correttamente: per quanto tempo?
- Quale dentifricio usare per lavarsi i denti?
- Come utilizzare correttamente il filo interdentale?
- Come lavare i denti ai bambini in modo facile e corretto
- Come lavarsi i denti in modo corretto [VIDEO]
- Come lavare i denti senza dentifricio
- Come lavarsi i denti senza spazzolino
- Come lavarsi i denti con l’apparecchio
- Come pulire l’apparecchio per i denti mobile e fisso
- Lingua infiammata (glossite): cause, sintomi, terapie, rimedi
- Lingua a carta geografica in bambini e adulti: stress, rimedi, quanto dura
- Glossite atrofica: cause, diagnosi, consigli e terapie
- Lingua bianca, impastata, spaccata: cause e quando è pericolosa
- In quali casi la lingua viene amputata?
- Lingua gialla con alito cattivo o mal gola: cause e rimedi
Dott. Emilio Alessio Loiacono
Medico Chirurgo
Direttore dello Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o unisciti al nostro gruppo Facebook o ancora seguici su Twitter, su Instagram, su Mastodon, su YouTube, su LinkedIn, su Tumblr e su Pinterest, grazie!
