
 La glicogenosi di tipo II (o di “tipo 2” o “malattia di Pompe” o “morbo di Pompe“) appartiene a un gruppo di malattie ereditarie caratterizzate dall’accumulo di glicogeno all’interno delle cellule o di alcuni organuli. In particolare, in questa malattia l’accumulo è a livello dei lisosomi, gli organuli intracellulari in cui avviene la degradazione di vari tipi di molecole. L’accumulo di glicogeno si verifica in vari tessuti: nel muscolo scheletrico, nel sistema nervoso, nel cuore e nel fegato, compromettendone la normale funzione. Alla base c’è il deficit di un enzima, chiamato acido alfaglucosidasi e deputato allo smaltimento del glicogeno nei lisosomi stessi.
La glicogenosi di tipo II (o di “tipo 2” o “malattia di Pompe” o “morbo di Pompe“) appartiene a un gruppo di malattie ereditarie caratterizzate dall’accumulo di glicogeno all’interno delle cellule o di alcuni organuli. In particolare, in questa malattia l’accumulo è a livello dei lisosomi, gli organuli intracellulari in cui avviene la degradazione di vari tipi di molecole. L’accumulo di glicogeno si verifica in vari tessuti: nel muscolo scheletrico, nel sistema nervoso, nel cuore e nel fegato, compromettendone la normale funzione. Alla base c’è il deficit di un enzima, chiamato acido alfaglucosidasi e deputato allo smaltimento del glicogeno nei lisosomi stessi.
Trasmissione
La glicogenosi di tipo 2 è dovuta a mutazioni nel gene GAA e si trasmette con modalità autosomica recessiva: l’allele alterato deve essere presente in coppia (omozigosi), cioè sono necessarie due copie dell’allele difettoso per far sì che la malattia si esprima, a prescindere dal sesso. Non basta un solo genitore portatore sano o malato, bensì entrambi i genitori devono essere portatori sani o malati. Il fenotipo quindi si esprime quando nel genotipo dell’individuo sono presenti entrambi gli alleli responsabili, fatto che spiega l’alta probabilità di sviluppare malattie genetiche in caso di incesto. Quindi:
- un individuo che possegga entrambi gli alleli alterati: è portatore ed è malato;
- un individuo che possegga solo un allele alterato: è portatore ma è sano;
- un individuo che non possegga nessun allele alterato: NON è portatore ed è sano.
Essere portatore sano vuol dire quindi NON avere la patologia ma possedere nel proprio genotipo un allele mutato, che può essere trasmesso alle generazioni successive.
Dalla combinazione delle possibili condizioni di genitori sani, malati e portatori sani, deriva la distribuzione probabilità che la malattia sia trasmessa ai figli:
- genitori malato-malato: la probabilità che il figlio/a nasca malato è del 100%;
- genitori sano-malato: la probabilità che il figlio/a nasca portatore sano è del 100%;
- genitori malato-portatore sano: la probabilità che il figlio/a nasca malato è del 50% e del 50% che nasca portatore sano;
- genitori sano-portatore sano: la probabilità che il figlio/a nasca sano è del 50% e del 50% che nasca portatore sano;
- genitori portatore-portatore: la probabilità che il figlio/a nasca portatore sano è del 50% mentre è del 25% che nasca sano o malato.
Se nessuno dei genitori ha un allele mutato, non c’è ovviamente alcuna trasmissione autosomica recessiva ed i figli saranno tutti sani e NON portatori dell’allele mutato.
Nell’immagine che segue, è raffigurata la tipica situazione in cui entrambi i genitori sono sani ma portatori dell’allele mutato:
- un figlio su quattro avrà entrambi gli alleli alterati e sarà malato ed ovviamente portatore;
- due figli su quattro avranno un allele normale ed uno alterato e saranno sani ma anche portatori;
- un figlio su quattro avrà entrambi gli alleli normali e sarà sano e NON portatore.

Le altre quattro situazioni possibili sono raffigurate nelle seguenti immagini:
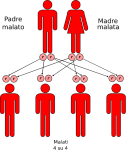
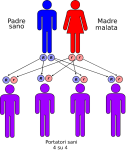
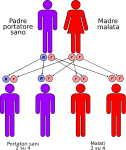
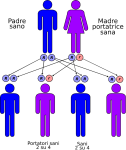
Sintomi e complicanze
Le manifestazioni della malattia sono variabili, a seconda dell’età di insorgenza. Le forme infantili sono le più gravi: caratterizzate da cardiomiopatia e grave debolezza muscolare, possono portare alla morte nel primo anno di vita. Le forme più tardive sono in genere più lievi, ma sono comunque caratterizzate da un indebolimento muscolare progressivo che può portare col tempo all’incapacità di camminare e all’insufficienza respiratoria.
Diagnosi
L’osservazione clinica porta a un sospetto diagnostico che può essere confermato dal dosaggio dell’enzima coinvolto in fibroblasti coltivati, in linfociti o in un campione di biopsia muscolare. È inoltre possibile effettuare l’analisi genetica (anche prenatale) con ricerca della mutazioni nel gene GAA. Attualmente questa malattia non rientra tra quelle oggetto dello screening neonatale esteso, se non in alcune Regioni (come per esempio la Toscana e il Veneto) dove è in corso una sperimentazione pilota: tuttavia le evidenze scientifiche emerse finora indicano come una diagnosi – e quindi una presa in carico – precoce possa contribuire a migliorare la qualità di vita di questi pazienti, per cui sarebbe auspicabile che in futuro anche la malattia di Pompe rientrasse tra quelle oggetto di screening alla nascita.
Terapia
E’ disponibile una terapia enzimatica sostitutiva, con iniezione periodica dell’enzima prodotto per via ricombinante (una volta ogni due settimane, per diverse ore). Questa terapia è molto efficace nelle forme infantili, mentre in alcune forme tardive può portare alla sola stabilizzazione della malattia e non a un efficace miglioramento. Inoltre, la somministrazione sistemica attraverso il sangue non permette di raggiungere efficacemente tutti gli organi colpiti. Per questo sono in fase di studio anche terapie alternative, come per esempio la terapia genica, o complementari, come l’impiego di farmaci chaperon in grado di migliorare gli effetti dalla terapia enzimatica sostitutiva.
Morbo di Pompe al cinema
La malattia di Pompe è citata nel film del 2010 “Misure straordinarie” diretto da Tom Vaughan ed interpretato da Harrison Ford e Brendan Fraser nella parte di John Crowley, basato su una storia vera, descritta nel libro “The Cure” scritto da Geeta Anand.
Leggi anche:
- Insufficienza epatica lieve, acuta e cronica: dieta e rischio di morte
- Cirrosi epatica e fegato: sintomi, dieta, diagnosi, terapia e prevenzione
- Funzionalità epatica; cos’è, cosa indica e come si misura
- Foetor hepaticus: causa, odore, definizione, significato
- Epatiti croniche: cosa sono, sintomi, diagnosi e cura
- Transaminasi alte, basse, cosa sono, cosa indicano e come si curano
- Cistifellea: cos’è, a cosa serve e dove si trova
- Dotto epatico comune, cistico e coledoco: anatomia del sistema biliare
- Sindrome post-colecistectomia: conseguenze dell’asportazione della cistifellea
- Si può vivere senza cistifellea?
- Vena porta e sistema portale: anatomia e funzioni della circolazione epatica
- Legatura/sclerosi delle varici esofagee: perché si esegue, quali sono i rischi?
- Elastografia epatica (FibroScan) per cirrosi: valori, preparazione all’esame, risultati
- Calcolosi colecisti: sintomi, dieta e terapie dei calcoli biliari
- Colecistite acuta e cronica litiasica e alitiasica: cause, terapia, dieta e rimedi naturali
- Dipendenza da alcol: come fare per smettere di bere alcolici e superalcolici
- L’alcol è una droga?
- Dove si trova il fegato ed a che serve?
- Differenza tra cirrosi e fibrosi
- Bile: dove si trova, a che serve e da cosa è composta?
- Bilirubina diretta e indiretta: ittero, significato, patologie collegate
- Ittero emolitico, colestatico, ostruttivo, neonatale: significato, occhi, cura
- Pelle gialla: differenza tra ittero e carotenodermia
- Anemia emolitica: farmaci, bilirubina, ittero, diagnosi di laboratorio
- Fegato ed epatociti: anatomia, funzioni e patologie in sintesi
- L’esofagogastroduodenoscopia: cos’è, preparazione, è dolorosa o pericolosa?
- Colangiopancreatografia retrograda (ERCP): cos’è, preparazione, è dolorosa o pericolosa?
- Fegato ed epatociti: anatomia, funzioni e patologie in sintesi
- Duodeno: anatomia e funzioni in sintesi
- Pancreas: anatomia e funzioni in sintesi
- Riconoscere i differenti tipi di vomito a seconda del colore
- Vomito: le cause più frequenti
- Vomito: rimedi naturali e cure farmacologiche (farmaci anti-emetici)
- Differenza tra intestino tenue e crasso
- Mal di pancia e di stomaco: da cosa può dipendere e quali sono le cure
- Mal di pancia forte: quando chiamare il medico?
- Differenza tra ulcera gastrica, duodenale, peptica ed esofagea
- Ulcera peptica: complicanze, cura, dieta, quando è pericolosa
- Ecografia renale: esecuzione, indicazioni, costo
- Ecografia alla vescica vuota o piena: preparazione, costo, svolgimento
- Feci dalla bocca: il vomito fecaloide
- Incontinenza fecale: dieta, Alzheimer, parto, cause e terapie
- Il mio alito odora di feci: cause, quando è pericoloso e rimedi
- Morbo di Crohn: cos’è, cause scatenanti, sintomi, cure e dieta
- Differenze tra ileo meccanico ed ileo paralitico: cause, sintomi e trattamenti
- Colite ulcerosa: cause, diagnosi, cura, dieta, cosa mangiare, rimedi
- Differenza tra colon irritabile, colite e colite spastica: sono la stessa cosa?
Lo Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o seguici su Twitter, su Instagram o su Pinterest, grazie!
