
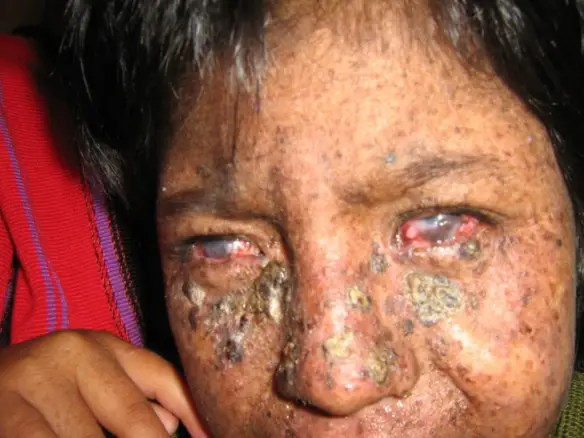
 Lo xeroderma pigmentoso (in inglese “xeroderma pigmentosum“) è una malattia genetica a ereditarietà autosomica recessiva caratterizzata da diminuita capacità nella riparazione dei danni al DNA, come quelli causati dalla luce ultravioletta (UV). Questa alterata capacità riparativa del materiale genico porta a sintomi e segni prevalentemente a carico della pelle, tra cui gravi scottature solari dopo una esposizione di solo pochi minuti al sole, lentiggini nelle aree esposte al sole, pelle secca, cambiamenti nella pigmentazione della pelle, predisposizione allo sviluppo di neoplasie e prematuro invecchiamento della pelle. Possono verificarsi anche cataratta, elevata fotosensibilità e problemi al sistema nervoso, come perdita dell’udito, scarsa coordinazione, perdita della funzione intellettiva e convulsioni. Le complicazioni includono un alto rischio di cancro della pelle, con circa la metà dei pazienti che ha un cancro della pelle all’età di 10 anni. Potrebbe esserci un rischio maggiore di altri tumori come i tumori al cervello. Gli individui con la malattia sono stati definiti “bambini della notte” o “bambini della luna” o “bambini vampiri“, proprio perché devono evitare il più possibile l’esposizione diretta ai raggi solari.
Lo xeroderma pigmentoso (in inglese “xeroderma pigmentosum“) è una malattia genetica a ereditarietà autosomica recessiva caratterizzata da diminuita capacità nella riparazione dei danni al DNA, come quelli causati dalla luce ultravioletta (UV). Questa alterata capacità riparativa del materiale genico porta a sintomi e segni prevalentemente a carico della pelle, tra cui gravi scottature solari dopo una esposizione di solo pochi minuti al sole, lentiggini nelle aree esposte al sole, pelle secca, cambiamenti nella pigmentazione della pelle, predisposizione allo sviluppo di neoplasie e prematuro invecchiamento della pelle. Possono verificarsi anche cataratta, elevata fotosensibilità e problemi al sistema nervoso, come perdita dell’udito, scarsa coordinazione, perdita della funzione intellettiva e convulsioni. Le complicazioni includono un alto rischio di cancro della pelle, con circa la metà dei pazienti che ha un cancro della pelle all’età di 10 anni. Potrebbe esserci un rischio maggiore di altri tumori come i tumori al cervello. Gli individui con la malattia sono stati definiti “bambini della notte” o “bambini della luna” o “bambini vampiri“, proprio perché devono evitare il più possibile l’esposizione diretta ai raggi solari.
Malattie progeroidi
Lo xeroderma pigmentoso fa parte di un gruppo di patologie dette “progeroidi”, che “determinano invecchiamento accelerato”, che comprende:
- progeria (sindrome di Benjamin Button);
- sindrome di Werner (progeria degli adulti);
- sindrome di Cockayne (sindrome di Weber-Cockayne);
- malattia di DeSanctis-Cacchione.
Quest’ultima rappresenta una variante dello xeroderma pigmentoso.
Etimologia
Questa malattia è così chiamata per via dei segni che provoca:
- pelle secca (xeroderma, dal greco ξηρός cioè “secco” e δέρμα, cioè “pelle”);
- pigmentoso (presenza di macchie scure irregolari sulla pelle).
Storia
Lo xeroderma pigmentoso è stato descritto per la prima volta nel 1870 dal medico e professore dermatologo ungherese Moritz Kaposi con l’aiuto di Ferdinand von Hebra. Nel 1882 Kaposi coniò il termine xeroderma pigmentosum per questa condizione, riferendosi alla sua caratteristica pelle secca e pigmentata. Kaposi è noto anche per aver descritto per primo alcune patologie quali il sarcoma di Kaposi e l’eczema herpeticum. Le anomalie nella riparazione del DNA nello xeroderma pigmentoso sono state portate all’attenzione della comunità scientifica dagli studi di James Cleaver nel 1968 che descrisse in un articolo il legame tra il danno al DNA indotto dai raggi UV, la riparazione difettosa del DNA e il cancro. Dalle ricerche sullo xeroderma pigmentoso alla fine del XX secolo è partita una riscrittura del concetto di cancerogenicità e di predisposizione genetica. La comprensione dell’eziologia dello xeroderma pigmentoso ha aiutato a comprendere i meccanismi biologici sottostanti molte altre malattie, non solo oncologiche.
Epidemiologia
Lo xeroderma pigmentoso ha un’incidenza globale di un caso ogni 250.000-1.000.000. Più specificatamente colpisce circa 1 persona su 370 in India, 1 persona su 20.000 in Giappone, 1 persona su 250.000 negli Stati Uniti e 1 su 430.000 in Europa. In Nord Africa e Pakistan è riportata una prevalenza di 1 ogni 40.000-100.000. Il gruppo più comune negli USA è quello C, mentre in Giappone è quello A. Piuttosto comuni anche quelli di gruppo D e lo Xeroderma pigmentoso gruppo variante. Come altre trasmissioni autosomiche recessive avviene più frequentemente quando sono più comuni le unioni tra consanguinei, quini la frequenza di questa malattia aumenta in regioni “chiuse” ed isolate, con scarsi fenomeni migratori. Si verifica ugualmente nei maschi e nelle femmine, senza predilezione per un sesso specifico.
Cause
Lo xeroderma pigmentoso ha causa genetica. L’anomalia è trasmessa con modalità autosomica recessiva, con mutazioni in una decina di geni specifici in grado di provocare la condizione (vedi paragrafo successivo). Normalmente, il danno al DNA che si verifica nelle cellule della pelle dall’esposizione alla luce UV e da altri elementi potenzialmente cancerogeni (come i raggi x) viene riparato dalla riparazione dell’escissione del nucleotide. Nelle persone con xeroderma pigmentoso, questo danno non viene riparato a causa del fatto che i geni che codificano per gli enzimi di riparazione dell’escissione nucleotidica (NER) sono mutati, portando a una riduzione dell’attività riparativa. Se non controllato, il danno causato dalla luce ultravioletta e da altri cancerogeni può causare mutazioni nel DNA delle singole cellule. Quando si formano più anomalie nel DNA, le cellule non funzionano correttamente e alla fine diventano cancerose o muoiono: ciò porta ai sintomi e segni dello xeroderma pigmentoso. Il processo alterato riguardante la riparazione del DNA danneggiato, è dato dalla perdita genetica della sub-unità riparatrice dell’enzima di trascrizione TFIIH della polimerasi-II negli umani. La TFIIH infatti, oltre a essere un enzima di trascrizione con funzione elicasica (dipendente dall’idrolisi di ATP) e oltre alla funzione fosforilatrice degli amminoacidi presenti nella coda carboxilo-terminale della pol-II, ha una funzione riparatrice del DNA danneggiato perché può reclutare un complesso di riparazione per escissione di nucleotidi; è proprio quest’ultima funzione che – come già detto – manca ai pazienti con xenoderma pigmentoso. I danni genici determinano l’alterazione delle seguenti proteine:
- proteina XPA: agisce durante il NER come un’impalcatura per l’assemblaggio di altre proteine di riparazione del DNA nei siti di danno al DNA per garantire un’appropriata escissione del danno;
- proteina XPB (ERCC3): viene impiegata per svolgere la doppia elica del DNA dopo che il danno al DNA è stato inizialmente riconosciuto. Le mutazioni nel gene XPB (ERCC3) possono portare a xeroderma pigmentoso o xeroderma pigmentoso combinato con la sindrome di Cockayne;
- proteina XPC: forma un complesso con la proteina RAD23B per formare il fattore di riconoscimento del danno iniziale nella riparazione dell’escissione del nucleotide genomico globale (GG-NER). Questo complesso riconosce un’ampia varietà di danni che destabilizzano termodinamicamente i duplex del DNA;
- proteina XPD (ERCC2): in combinazione con il complesso di trascrizione/riparazione XPB contenente elicasi TFIIH, viene impiegata per svolgere il duplex del DNA dopo che il danno è stato inizialmente riconosciuto. Le mutazioni nel gene XPD (ERCC2) causano una varietà di sindromi; xeroderma pigmentoso, tricotiodistrofia (TTD) o una combinazione di xeroderma pigmentoso e sindrome di Cockayne. Sia la tricotiodistrofia che la sindrome di Cockayne mostrano caratteristiche dell’invecchiamento prematuro, suggerendo un’associazione tra la riparazione del DNA carente e l’invecchiamento prematuro;
- proteina XPE: è una proteina eterodimerica composta da due subunità. La subunità più grande DDB1 funziona principalmente come componente principale dei complessi di ubiquitina ligasi E3 basati su CUL4A e CUL4B. I substrati che sono ubiquitinati da questi complessi includono proteine impiegate nella riparazione del DNA;
- proteina XPF (ERCC4): insieme alla proteina ERCC1 forma un complesso solitamente designato ERCC1-XPF. Questo complesso separa l’elica del DNA per una breve distanza su entrambi i lati del sito del danno. Agisce quindi come un’endonucleasi per incidere il filamento di DNA danneggiato sul lato 5′ del sito danneggiato. Le cellule mutanti con ERCC1-XPF carente non sono solo difettose nel NER, ma anche nella riparazione delle rotture del doppio filamento e dei legami incrociati tra i filamenti;
- proteina XPG: è un’endonucleasi che incide il DNA durante il NER sul lato 3′ del nucleotide danneggiato. Le mutazioni nel gene XPG (ERCC5) possono portare a XP da solo, o in combinazione con la sindrome di Cockayne (CS), o in combinazione con la sindrome cerebro-oculo-facciale letale infantile.
Classificazione
In base ai geni coinvolti, esistono sette sottogruppi principali di xeroderma pigmentoso:
| Tipo | Gene | Loci | Nome |
| Tipo A, I, XPA | XPA | 9q22.3 | Xeroderma pigmentoso gruppo A |
| Tipo B, II, XPB | XPB | 2q21 | Xeroderma pigmentoso gruppo B |
| Tipo C, III, XPC | XPC | 3p25 | Xeroderma pigmentoso gruppo C |
| Tipo D, IV, XPD | XPD ERCC6 | 19q13.2-q13.3 , 10q11 | Xeroderma pigmentoso gruppo D o malattia di DeSanctis-Cacchione |
| Tipo E, V, XPE | DDB2 | 11p12-p11 | Xeroderma pigmentoso gruppo E |
| Tipo F, VI, XPF | ERCC4 | 16p13.3-p13.13 | Xeroderma pigmentoso gruppo F |
| Tipo G, VII, XPG | RAD2 ERCC5 | 13q33 | Xeroderma pigmentoso gruppo G |
| Tipo V, XPV | POLH | 6p21.1-p12 | Xeroderma pigmentoso gruppo variante |
- XPA è una proteina dalla funzione incerta, il suo compito sembra essere quello di verificare la corretta posizione di altre proteine nel contesto del danno al DNA così da poter farlo incidere da specifiche nucleasi.
- XPB e XPD sono subunità con attività elicasica del fattore di trascrizione TFIIH.
- XPC riconosce il sito in cui è danneggiato il DNA insieme ad HR23B.
- XPE è una subunità proteica di un eterodimero che si lega specificatamente a DNA danneggiato da raggi UV (dimeri di pirimidina) e stimola la reazione di incisione.
- XPF e XPG sono nucleasi.
- XPV/POLH codifica per la DNA Pol η.
Trasmissione autosomica recessiva
Lo xeroderma pigmentoso è una malattia autosomica recessiva. Una malattia è detta a trasmissione autosomica recessiva quando l’allele alterato deve essere presente in coppia (omozigosi), cioè sono necessarie due copie dell’allele difettoso per far sì che la malattia si esprima, a prescindere dal sesso. Non basta un solo genitore portatore sano o malato, bensì entrambi i genitori devono essere portatori sani o malati. Il fenotipo quindi si esprime quando nel genotipo dell’individuo sono presenti entrambi gli alleli responsabili, fatto che spiega l’alta probabilità di sviluppare malattie genetiche in caso di incesto. Quindi:
- un individuo che possegga entrambi gli alleli alterati: è portatore ed è malato;
- un individuo che possegga solo un allele alterato: è portatore ma è sano;
- un individuo che non possegga nessun allele alterato: NON è portatore ed è sano.
Essere portatore sano vuol dire quindi NON avere la patologia ma possedere nel proprio genotipo un allele mutato, che può essere trasmesso alle generazioni successive.
Dalla combinazione delle possibili condizioni di genitori sani, malati e portatori sani, deriva la distribuzione probabilità che la malattia sia trasmessa ai figli:
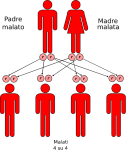
- genitori malato-malato: la probabilità che il figlio/a nasca malato è del 100%;
- genitori sano-malato: la probabilità che il figlio/a nasca portatore sano è del 100%;
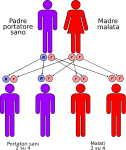
- genitori malato-portatore sano: la probabilità che il figlio/a nasca malato è del 50% e del 50% che nasca portatore sano;
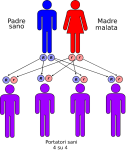
- genitori sano-portatore sano: la probabilità che il figlio/a nasca sano è del 50% e del 50% che nasca portatore sano;
- genitori portatore-portatore: la probabilità che il figlio/a nasca portatore sano è del 50% mentre è del 25% che nasca sano o malato.
Se nessuno dei genitori ha un allele mutato, non c’è ovviamente alcuna trasmissione autosomica recessiva ed i figli saranno tutti sani e NON portatori dell’allele mutato.
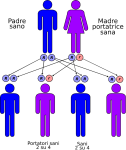
Nell’immagine che segue, è raffigurata la tipica situazione in cui entrambi i genitori sono sani ma portatori dell’allele mutato:
- un figlio su quattro avrà entrambi gli alleli alterati e sarà malato ed ovviamente portatore;
- due figli su quattro avranno un allele normale ed uno alterato e saranno sani ma anche portatori;
- un figlio su quattro avrà entrambi gli alleli normali e sarà sano e NON portatore.

Le altre quattro situazioni possibili sono raffigurate nelle seguenti immagini:
Sintomi e segni
Lo xeroderma pigmentoso mostra un’elevata variabilità clinica correlata sia al gene mutato che al tipo di mutazione presente. I sintomi e segni più comuni sono:
- gravi scottature solari se esposti anche solo a piccole quantità di luce solare. Questi si verificano spesso durante la prima esposizione di un bambino alla luce solare;
- sviluppo di molte lentiggini in tenera età;
- escrescenze superficiali (cheratosi solari);
- tumori della pelle sulle aree esposte (mani, braccia, viso);
- fotosensibilità: gli occhi sono dolorosamente sensibili al sole e possono facilmente irritarsi, iniettarsi di sangue e annebbiarsi;
- vesciche o lentiggini alla minima esposizione al sole;
- telangiectasia (“vene del ragno”);
- crescita limitata dei peli su petto e gambe;
- pelle squamosa;
- xeroderma (pelle secca);
- macchie scure irregolari sulla pelle;
- ulcerazioni corneali;
- pseudo-poichilodermia gradualepredominante nelle aree esposte alla luce solare o scarsamente protette.
Esistono differenze cliniche in base al tipo di malattia. I pazienti del gruppo A tendono a sviluppare problemi neurologici nella prima infanzia mentre quelli di gruppo D nella tarda infanzia. I pazienti di gruppo C invece non sviluppano problemi di questo tipo. I sintomi e i segni clinici presentano una fotosensibilità estrema, cheratosi, gran numero di lentiggini, comparsa di tumori cutanei. Nel gruppo D si mostrano bambini di bassa statura, capelli fragili, gran numero di lentiggini e scarsa intelligenza, nel gruppo C si arriva all’atassia.
Età di esordio
La malattia è congenita, cioè già presente alla nascita, tuttavia i primi segni cutanei compaiono tra l’età di 1 e 2 anni, quando alla nascita la pelle appariva normale. Il primo segno è spesso una reazione anormalmente grave dopo un’esposizione anche minima al sole: eritema insolito per intensità e durata nelle aree esposte.
Diagnosi
La diagnosi di xeroderma pigmentoso si basa sui sintomi e segni e sulla ipersensibilità nei confronti delle radiazioni UV. Viene confermata dai test cellulari e genetici che individuano i difetti della riparazione del DNA.
Diagnosi differenziale
La diagnosi differenziale si pone con la tricotiodistrofia, la sindrome di Cockayne, cerebro-oculo-facio-scheletrica, e di Rothmund-Thomson, e la protoporfiria eritropoietica.
Diagnosi prenatale
È possibile la diagnosi prenatale mediante il dosaggio della UDS (test della sintesi non programmata del DNA) nei villi coriali o negli amniociti in coltura.
Terapie
Non esiste una cura specifica lo xeroderma pigmentoso. Viene trattato indirettamente, prevenendo l’esposione al sole e gestendo i sintomi e segni associati per migliorare la qualità della vita.
I sintomi possono essere evitati o controllati evitando completamente l’esposizione alla luce solare, rimanendo all’interno o indossando indumenti protettivi e utilizzando la protezione solare quando si è all’aperto. Nei casi più gravi di xeroderma pigmentoso, anche quantità minime raggi UV, ad esempio provenienti da finestre coperte con tende o lampadine fluorescenti, possono essere molto pericolose e scatenare sintomi e segni.
Gli effetti della malattia possono isolare il paziente ed indurlo a depressione e pensieri suicidari: un trattamento psicoterapico potrebbe aiutare la situazione.
La cheratosi può anche essere trattata utilizzando la crioterapia o il fluorouracile. Lesioni cutanee piccole e precancerose come le cheratosi attiniche possono essere rimosse con azoto liquido. Le aree più grandi di pelle danneggiata dal sole possono essere trattate con fluorouracile o imiquimod topico. In rari casi, il peeling del dermatoma o la dermoabrasione vengono utilizzate per rimuovere gli strati epidermici superficiali danneggiati. I cancri della pelle possono essere trattati utilizzando protocolli standard, tra cui la escissione chirurgica o la chemiochirurgia.
L’isotretinoina o l’acitretina orali ad alte dosi possono essere utilizzate per prevenire nuovi tumori. I cancri delle palpebre, della congiuntiva e della cornea vengono solitamente trattati chirurgicamente. Il trapianto di cornea può essere necessario per soggetti con cheratite grave e opacità corneale.
Il 10 settembre 2020, Clinuvel Pharmaceuticals ha annunciato che stava studiando l’uso del suo farmaco di punta approvato dalla FDA Scenesse come potenziale trattamento per aumentare l’esposizione alla luce indolore per i pazienti con xeroderma pigmentoso.
Prevenzione
La prevenzione degli effetti più gravi dello xeroderma pigmentoso, prevede:
- protezione dalla luce ultravioletta;
- frequenti esami della pelle e degli occhi;
- rimozione di tessuto canceroso negli stadi iniziali del tumore;
- alimentazione ricca di vitamine;
- esami neurologici periodici.
Rischio di tumore
Gli individui con xeroderma pigmentoso hanno circa 10.000 volte più probabilità di sviluppare il cancro della pelle (non melanoma) rispetto agli individui senza il disturbo, mentre il rischio di sviluppare melanoma prima dei 20 anni è maggiore di 2.000 volte. Carcinomi a cellule basali e altri tumori maligni della pelle si verificano frequentemente in giovane età (spesso si verifica prima che il bambino abbia 5 anni).
Prognosi e mortalità
L’aspettativa di vita media di un individuo con qualsiasi tipo di xeroderma pigmentoso e nessun sintomo neurologico è di circa 37 anni; se sono presenti sintomi neurologici l’aspettativa si abbassa a circa 29 anni. Meno del 40% degli individui con la malattia sopravvive oltre i 20 anni. Negli Stati Uniti, la probabilità che gli individui con il disturbo sopravvivano fino a 40 anni di età può raggiungere il 70% se non sono mai stati esposti alla luce solare in vita loro. In India, molti pazienti con xeroderma pigmentoso muoiono in tenera età a causa di melanoma, un cancro della pelle molto aggressivo e spesso fatale. Se una persona viene diagnosticata precocemente, non presenta sintomi neurologici gravi e adotta misure precauzionali per evitare completamente l’esposizione ai raggi UV e alla luce solare, potrebbe essere in grado di sopravvivere fino a quasi 50 anni. L’aspettativa di vita aumenta in particolare nei pazienti che:
- evitano la luce solare;
- si sottopongono a follow-up regolari per valutare e curare i tumori della pelle;
- non hanno disturbi neurologici.
Il melanoma maligno metastatico e il carcinoma a cellule squamose sono le due cause più comuni di morte nei pazienti con xeroderma pigmentoso.
Curiosità
Lo xeroderma pigmentoso è stato un elemento della trama in diverse opere di fantasia. Children of Darkness, un film drammatico muto tedesco uscito in due parti rispettivamente nell’anno 1921 e 1922, è stato tra alcuni dei film inizialmente popolari realizzati su xeroderma pigmentoso. Altri film, come il film drammatico americano del 1964 Della, con Joan Crawford, Paul Burke, Charles Bickford e Diane Baker, diretto da Robert Gist, si basava anche su questa malattia. The Dark Side of the Sun, un film drammatico americano-jugoslavo del 1988, è stato diretto da Božidar Nikolić e interpretato da Brad Pitt per il suo primo ruolo da protagonista da giovane in cerca di una cura per il disturbo. Il popolare film horror psicologico del 2001 The Others, con Nicole Kidman, presenta due bambini, Anne e Nicholas, che devono evitare tutta la luce solare a causa di una malattia rara caratterizzata dalla fotosensibilità. Un film per la televisione della CBS andato in onda nel 1994, Children of the Dark, era basato sulla storia della coppia nella vita reale Jim e Kim Harrison, le cui due figlie hanno lo xeroderma pigmentoso. Il film drammatico francese del 2011 The Moon Child è basato su un bambino di 13 anni con uno xeroderma pigmentoso che gli impedisce di esporsi alla luce del giorno. Il film d’amore del 2018 Midnight Sun, basato su un film giapponese del 2006, A Song to the Sun, racconta la storia di una ragazza di nome Katie Price con XP e l’impatto della sua malattia sulla sua vita e sulle sue relazioni, in seguito alla storia dell’esposizione accidentale di Price alla luce solare e la successiva degenerazione neurologica.
Articoli sulle malattie progeroidi:
- Progeria (sindrome di Benjamin Button): cause, trasmissione, età di esordio e sintomi
- Progeria (sindrome di Benjamin Button): diagnosi, terapie e aspettativa di vita
- Sindrome di Werner (progeria degli adulti): cause, trasmissione, sintomi, diagnosi, cure, prognosi
- Sindrome di Cockayne (sindrome di Weber-Cockayne): cause, sintomi, diagnosi, cure, prognosi
- Malattia di DeSanctis-Cacchione: cause, sintomi, diagnosi e terapie
Leggi anche:
- Progeria: addio a Sam Berns, il bambino che invecchiava troppo in fretta
- Malattia di Charcot-Marie-Tooth: cause, sintomi, diagnosi, terapia
- Le malattie genetiche più diffuse al mondo
- Differenza tra autosomica dominante e recessiva con esempi
- Sindrome di Turner: cariotipo, cause, sintomi e segni caratteristici
- Sindrome di Klinefelter: cariotipo, cause, sintomi e cura
- Sindrome di Down: cause, sintomi in gravidanza e nei neonati
- Fibrosi cistica polmonare: cos’è, sintomi in neonati e bambini, cure
- Malattia di Huntington: cos’è, ereditarietà, come si trasmette, età di insorgenza
- Anemia falciforme: cosa significa, cause, sintomi e cure
- Differenze tra la distrofia muscolare di Duchenne e di Becker
- Talassemia: cos’è, sintomi, cure, differenti tipi ed alimentazione
- Celiachia: cos’è il glutine, in quali alimenti è contenuto ed in quali no?
- Sindrome di Noonan: cause, sintomi nel neonato, aspettative di vita
- Sindrome di Bloom: cause, sintomi, diagnosi e terapia
- Bimba nata con organi fuori dal corpo, glieli avvolgono con una pellicola
- Münchhausen per procura: perché questa mamma tortura il proprio bambino?
- Spina bifida e difetti di chiusura del tubo neurale nel feto: trasmissione, prevenzione, diagnosi e cura
- Mielomeningocele, spina bifida e schisi vertebrale: complicanze, cura, prevenzione
- Encefalocele: cause, sintomi, diagnosi, cura e prevenzione
- Cos’è un cromosoma ed a che serve?
- Cos’è un gene ed a che serve?
- Differenza tra malattia genetica, ereditaria e congenita
Dott. Emilio Alessio Loiacono
Medico Chirurgo
Direttore dello Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o unisciti al nostro gruppo Facebook o ancora seguici su Twitter, su Instagram, su Mastodon, su Tumblr e su Pinterest, grazie!
