
 La sindrome di Cockayne (anche chiamata “sindrome di Weber-Cockayne” o “sindrome di Neill-Dingwall“; in inglese “Cockayne syndrome” o “Neill-Dingwall syndrome” o “Weber-Cockayne sindrome“), è una malattia neurodegenerativa autosomica recessiva rara e fatale caratterizzata da ritardo della crescita, alterato sviluppo del sistema nervoso, sensibilità anormale alla luce solare (fotosensibilità), disturbi dell’occhio e invecchiamento precoce. I pazienti passano rapidamente dall’infanzia alla vecchiaia, saltando di fatto adolescenza ed età adulta. È associata a un gruppo di disturbi chiamati leucodistrofie, che sono condizioni caratterizzate dalla degradazione della sostanza bianca del sistema nervoso. La causa della sindrome è un difetto in un meccanismo di riparazione del DNA, ma a differenza di altri difetti di riparazione del DNA, i pazienti con CS non sono predisposti al cancro o alle infezioni. La sindrome di Cockayne è una malattia rara ma distruttiva che di solito provoca la morte entro la prima o la seconda decade di vita. La mutazione di geni specifici nella sindrome di Cockayne è nota, ma gli effetti diffusi e la sua relazione con la riparazione del DNA devono ancora essere ben compresi. Dal momento che la sindrome di Cockayne viene anche chiamata sindrome di Weber-Cockayne, ricordiamo al lettore che non deve essere confusa con la sindrome di Weber o la sindrome di Sturge-Weber o la sindrome di Klippel-Trénaunay-Weber o con la sindrome di Parkes Weber.
La sindrome di Cockayne (anche chiamata “sindrome di Weber-Cockayne” o “sindrome di Neill-Dingwall“; in inglese “Cockayne syndrome” o “Neill-Dingwall syndrome” o “Weber-Cockayne sindrome“), è una malattia neurodegenerativa autosomica recessiva rara e fatale caratterizzata da ritardo della crescita, alterato sviluppo del sistema nervoso, sensibilità anormale alla luce solare (fotosensibilità), disturbi dell’occhio e invecchiamento precoce. I pazienti passano rapidamente dall’infanzia alla vecchiaia, saltando di fatto adolescenza ed età adulta. È associata a un gruppo di disturbi chiamati leucodistrofie, che sono condizioni caratterizzate dalla degradazione della sostanza bianca del sistema nervoso. La causa della sindrome è un difetto in un meccanismo di riparazione del DNA, ma a differenza di altri difetti di riparazione del DNA, i pazienti con CS non sono predisposti al cancro o alle infezioni. La sindrome di Cockayne è una malattia rara ma distruttiva che di solito provoca la morte entro la prima o la seconda decade di vita. La mutazione di geni specifici nella sindrome di Cockayne è nota, ma gli effetti diffusi e la sua relazione con la riparazione del DNA devono ancora essere ben compresi. Dal momento che la sindrome di Cockayne viene anche chiamata sindrome di Weber-Cockayne, ricordiamo al lettore che non deve essere confusa con la sindrome di Weber o la sindrome di Sturge-Weber o la sindrome di Klippel-Trénaunay-Weber o con la sindrome di Parkes Weber.
Malattie progeroidi
La sindrome di Cockayne fa parte di un gruppo di patologie dette “progeroidi”, che “determinano invecchiamento accelerato”, che comprende:
- progeria (sindrome di Benjamin Button);
- sindrome di Werner (progeria degli adulti);
- xeroderma pigmentoso;
- malattia di DeSanctis-Cacchione.
Eponimo
La sindrome di Cockayne deve il suo nome al pediatra inglese Edward Alfred Cockayne (1880 -1956) che la descrisse per primo nel 1936 e la rianalizzò approfonditamente nel 1946. La sindrome di Cockayne viene anche chiamata “sindrome di Neill-Dingwall” o “sindrome di Weber-Cockayne” in onore di Mary Dingwall, Catherine Neill e Frederick Parkes Weber che la studiarono in seguito. In particolare Mary M. Dingwall e Catherine A. Neill descrissero il caso di due fratelli con la sindrome di Cockayne e notarono che probabilmente si trattava della stessa malattia descritta da Cockayne. Nel loro articolo, i due hanno contribuito ai segni della malattia attraverso la scoperta di calcificazioni nel cervello. Hanno anche confrontato la sindrome di Cockayne con la “progeria” (anche chiamata “sindrome di Hutchinson-Gilford” o “sindrome di Hutchinson”) a causa dell’invecchiamento avanzato che caratterizza entrambi i disturbi.
Classificazione
La sindrome di Cockayne viene classificata in tre tipi di cui il secondo è il più grave mentre il terzo è il più lieve:
- Sindrome di Cockayne tipo I (o “tipo A”), la forma “classica”, è caratterizzata da una normale crescita fetale con insorgenza di anomalie nei primi due anni di vita. La vista e l’udito diminuiscono gradualmente. Il sistema nervoso centrale e periferico degenera progressivamente fino alla morte nella prima o seconda decade di vita per grave degrado neurologico. L’atrofia corticale è meno grave in questo primo tipo di sindrome di Cockayne.
- Sindrome di Cockayne tipo II (o “tipo B”) è presente dalla nascita (congenita) ed è molto più grave del tipo I. Implica uno sviluppo neurologico minimo dopo la nascita. La morte di solito si verifica all’età di sette anni. Questo tipo specifico è stato anche designato come sindrome cerebro-oculo-facciale-scheletrica (in inglese “cerebro-oculo-facio-skeletal syndrome”, da cui l’acronimo COFS) o sindrome di Pena-Shokeir di tipo II. La sindrome COFS è chiamata così a causa degli effetti che ha sul cervello, sugli occhi, sul viso e sul sistema scheletrico, poiché la malattia provoca spesso atrofia cerebrale, cataratta, perdita di grasso nel viso e marcata osteoporosi. La sindrome COFS può essere ulteriormente suddivisa in diverse condizioni: COFS di tipo I, II e III (associata a xeroderma pigmentoso) e IV. Tipicamente i pazienti con questa forma ad esordio precoce del disturbo mostrano un danno cerebrale più grave, inclusa una ridotta mielinizzazione della sostanza bianca e calcificazioni più diffuse, anche nella corteccia e nei gangli della base.
- Sindrome di Cockayne tipo III (o “tipo C”), caratterizzato da esordio tardivo, è in genere più lieve rispetto ai tipi I e II. Spesso i pazienti con tipo III vivranno fino all’età adulta. Prende anche il nome di sindrome dello xeroderma pigmentoso-Cockayne (XP-CS) e si verifica quando un individuo ha anche lo xeroderma pigmentoso, un’altra malattia di riparazione del DNA. Alcuni sintomi di ciascuna malattia sono espressi. Ad esempio, sono presenti le lentiggini e le anomalie del pigmento caratteristiche di XP. Si osservano il disturbo neurologico, la spasticità e il sottosviluppo degli organi sessuali caratteristici della CS. Tuttavia, l’ipomielinizzazione e le caratteristiche facciali dei tipici pazienti con CS non sono presenti.
Eziopatologia
Se nel corpo si verifica iperossia o eccesso di ossigeno, il metabolismo cellulare produce diverse forme di ossigeno altamente reattive chiamate radicali liberi. Ciò può causare danni ossidativi ai componenti cellulari, incluso il DNA. Nelle cellule normali, il nostro corpo ripara le sezioni danneggiate, invece, nel caso di questa malattia, a causa di difetti di trascrizione genica, il meccanismo dei bambini per sintetizzare le proteine necessarie all’organismo per riparare i danni non funziona normalmente. Per valutare l’entità del danno che il malfunzionamento di questi meccanismi di riparazione comportano, ricordiamo che i radicali liberi possono causare danni ossidativi ai componenti cellulari compreso il DNA e che in una cellula umana media, ogni giorno si verificano diverse decine di migliaia di lesioni nel DNA derivate dal danno ossidativo. Ogni lesione, una sezione danneggiata del DNA, deve essere tagliata e il DNA riparato per preservare la sua normale funzione. Il DNA non riparato può perdere la sua capacità di codificare le proteine e possono verificarsi anche mutazioni. Queste mutazioni possono attivare oncogeni o silenziare i geni oncosoppressori. Secondo la ricerca, il danno ossidativo ai geni attivi non è riparato preferenzialmente e, nei casi più gravi, la riparazione è rallentata in tutto il genoma. Il conseguente accumulo di danno ossidativo potrebbe compromettere le normali funzioni del DNA e potrebbe anche provocare l’innesco di un programma di morte cellulare (apoptosi). I bambini con questa malattia non riparano il danno ossidativo ed il conseguente accumulo di danno ossidativo è alla base dei sintomi e segni di questa sindrome e porta a un invecchiamento precoce.
Cause
La sindrome di Cockayne ha cause genetiche. In base al tipo, i geni interessati sono:
| Tipo | Gene | |
|---|---|---|
| I | ERCC8 (CSA) | |
| II | ERCC6 (CSB) | |
| III | sconosciuto |
Le mutazioni nel gene ERCC8 (noto anche come CSA) o nel gene ERCC6 (noto anche come CSB) sono la causa della sindrome di Cockayne di tipo I e II. Le mutazioni nella mutazione del gene ERCC6 costituiscono circa il 70% dei casi mentre quelle del gene ERCC8 sono il 20% circa dei casi totali. Le proteine prodotte da questi geni hanno il compito di riparare il DNA danneggiato attraverso il meccanismo di riparazione accoppiato alla trascrizione, in particolare il DNA nei geni attivi. Il danno al DNA è causato dai raggi ultravioletti della luce solare, dalle radiazioni o dai radicali liberi nel corpo. Una cellula normale può riparare il danno al DNA prima che si accumuli. Se il gene ERCC6 o ERCC8 è alterato (come nella sindrome di Cockayne), il danno al DNA riscontrato durante la trascrizione non viene riparato, causando lo stallo dell’RNA polimerasi in quella posizione, interferendo con l’espressione genica. Man mano che il danno al DNA non riparato si accumula, l’espressione genica progressivamente più attiva viene impedita, portando a cellule malfunzionanti o morte cellulare (apoptosi), che probabilmente contribuisce ai segni della sindrome di Cockayne come l’invecchiamento precoce e l’ipomielinizzazione neuronale.
Trasmissione autosomica recessiva
La sindrome di Cockayne è una malattia genetica che si trasmette con modalità autosomica recessiva. Una malattia è detta a trasmissione autosomica recessiva quando l’allele alterato deve essere presente in coppia (omozigosi), cioè sono necessarie due copie dell’allele difettoso per far sì che la malattia si esprima, a prescindere dal sesso. Non basta un solo genitore portatore sano o malato, bensì entrambi i genitori devono essere portatori sani o malati. Il fenotipo quindi si esprime quando nel genotipo dell’individuo sono presenti entrambi gli alleli responsabili, fatto che spiega l’alta probabilità di sviluppare malattie genetiche in caso di incesto. Quindi:
- un individuo che possegga entrambi gli alleli alterati: è portatore ed è malato;
- un individuo che possegga solo un allele alterato: è portatore ma è sano;
- un individuo che non possegga nessun allele alterato: NON è portatore ed è sano.
Essere portatore sano vuol dire quindi NON avere la patologia ma possedere nel proprio genotipo un allele mutato, che può essere trasmesso alle generazioni successive.
Dalla combinazione delle possibili condizioni di genitori sani, malati e portatori sani, deriva la distribuzione probabilità che la malattia sia trasmessa ai figli:
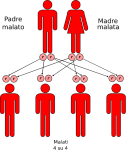
- genitori malato-malato: la probabilità che il figlio/a nasca malato è del 100%;
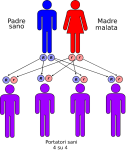
- genitori sano-malato: la probabilità che il figlio/a nasca portatore sano è del 100%;
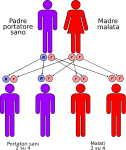
- genitori malato-portatore sano: la probabilità che il figlio/a nasca malato è del 50% e del 50% che nasca portatore sano;
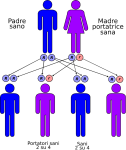
- genitori sano-portatore sano: la probabilità che il figlio/a nasca sano è del 50% e del 50% che nasca portatore sano;
- genitori portatore-portatore: la probabilità che il figlio/a nasca portatore sano è del 50% mentre è del 25% che nasca sano o malato.
Se nessuno dei genitori ha un allele mutato, non c’è ovviamente alcuna trasmissione autosomica recessiva ed i figli saranno tutti sani e NON portatori dell’allele mutato.
Nell’immagine che segue, è raffigurata la tipica situazione in cui entrambi i genitori sono sani ma portatori dell’allele mutato:
- un figlio su quattro avrà entrambi gli alleli alterati e sarà malato ed ovviamente portatore;
- due figli su quattro avranno un allele normale ed uno alterato e saranno sani ma anche portatori;
- un figlio su quattro avrà entrambi gli alleli normali e sarà sano e NON portatore.

Le altre quattro situazioni possibili sono raffigurate nelle seguenti immagini:
Sintomi e segni
Le persone con questa sindrome hanno dimensioni della testa più piccole del normale (microcefalia), sono di bassa statura (nanismo), i loro occhi sembrano infossati e hanno un aspetto “invecchiato”. Il bambino nasce di solito sottopeso e manifesta una serie di ritardi nella crescita. Dopo l’anno d’età manifesta forti tremori che ostacolano le funzioni motorie. La cataratta è precoce. I soggetti hanno spesso arti lunghi con contratture articolari (incapacità di rilassare il muscolo in corrispondenza di un’articolazione), schiena curva (cifosi) e possono essere molto magri (cachetici), a causa della perdita di grasso sottocutaneo. Il reflusso gastroesofageo è frequente. Il mento piccolo, le orecchie grandi e il naso sottile e appuntito danno spesso al paziente un aspetto invecchiato rispetto alla sua vera età. Anche la pelle delle persone con sindrome di Cockayne è frequentemente colpita: iperpigmentazione, varici o vene varicose (telangiectasia) e grave sensibilità alla luce solare sono comuni. Spesso i pazienti con la sindrome di Cockayne si ustionano gravemente o si formano vesciche con un’esposizione al calore molto ridotta. Gli occhi dei pazienti possono essere colpiti in vari modi e le anomalie oculari sono comuni. Frequente, oltre la cataratta, anche l’opacità della cornea (opacità corneale). Possono verificarsi la perdita e il danneggiamento dei nervi del nervo ottico, causando atrofia ottica. Il nistagmo, o movimento oculare involontario, e le pupille che non si dilatano dimostrano una perdita di controllo del movimento muscolare volontario e involontario. Un segno tipico è anche una pigmentazione retinica “sale e pepe”.
Diagnosi
La diagnosi della sindrome di Cockayne si basa sui sintomi e segni caratteristici della sindrome e si conferma con test genetici ed un test specifico per la riparazione del DNA, che misura il recupero dell’RNA dopo l’esposizione ai raggi UV. Nei pazienti con sindrome di Cockayne, le cellule irradiate dai raggi UV mostrano una ridotta sintesi di DNA e RNA. Gli studi di laboratorio e di diagnostica per immagini sono utili per la diagnosi differenziale. Ad esempio, la radiografia scheletrica, i test endocrinologici e gli studi sulla rottura cromosomica possono aiutare a escludere i disturbi inclusi nella diagnosi differenziale. La TC cerebrale nei pazienti con sindrome di Cockayne può rivelare calcificazioni e atrofia corticale. Studi di imaging rivelano una diffusa assenza delle guaine mieliniche dei neuroni nella sostanza bianca del cervello e un’atrofia generale della corteccia. Calcificazioni sono state trovate anche nel putamen, un’area del proencefalo che regola i movimenti e aiuta in alcune forme di apprendimento, insieme alla corteccia. L’atrofia dell’area centrale del cervelletto riscontrata nei pazienti con sindrome di Cockayne potrebbe anche comportare la mancanza di controllo muscolare, in particolare involontario, e la postura scorretta tipicamente osservata.
Diagnosi prenatale
È possibile la valutazione prenatale. La coltura cellulare del liquido amniotico preleviato con amniocentesi viene utilizzata per dimostrare che le cellule fetali sono carenti nella sintesi dell’RNA dopo l’irradiazione UV. Si raccomanda la consulenza genetica per i genitori, poiché il disturbo ha una probabilità del 25% di essere trasmesso a futuri figli e anche i test prenatali sono una possibilità. Un altro aspetto importante è la prevenzione delle recidive della sindrome di Cockayne negli altri fratelli. L’identificazione dei difetti genetici coinvolti consente di offrire consulenza genetica e test diagnostici prenatali ai genitori che hanno già un figlio affetto.
Diagnosi differenziale
La sindrome di Cockayne deve essere distinta da altre malattie progeroidi. Nonostante sia associato a geni coinvolti nella riparazione dell’escissione nucleotidica, a differenza dello xeroderma pigmentoso, la sindrome di Cockayne non è associata ad un aumentato rischio di cancro.
Trattamento
Non esiste una cura specifica la sindrome di Cockayne. Viene trattata indirettamente, gestendo le malattie associate e alleviando i sintomi per migliorare la qualità della vita. Il trattamento di solito prevede la terapia fisica e piccoli interventi chirurgici agli organi colpiti, come la rimozione della cataratta. Si consiglia inoltre di indossare una crema solare ad alto fattore e indumenti protettivi perché i pazienti con sindrome di Cockayne sono molto sensibili ai raggi UV. Anche un’alimentazione ottimale ed una attività sportiva regolare possono aiutare a rallentare la malattia. Importante evitare qualsiasi comportamento che possa aumentare le possibilità di danno cellulare: non fumare, non bere alcolici, non assumere droghe, evitare ambienti e professioni che espongano ad inquinanti, evitare se possibile l’esposizione a radiazioni (ad esempio radiografie), evitare cibi fritti.
Prognosi e mortalità
La prognosi per le persone con sindrome di Cockayne è purtroppo sfavorevole, poiché la morte si verifica in genere mediamente entro l’età di 12 anni, tuttavia la prognosi per la sindrome di Cockayne varia moltissimo in base al tipo di malattia. Come già anticipato esistono tre tipi di sindrome di Cockayne in base alla gravità e all’insorgenza dei sintomi, tuttavia, le differenze tra i tipi non sono sempre nette e alcuni ricercatori ritengono che i segni e i sintomi riflettano uno spettro invece di tipi distinti:
- la sindrome di Cockayne di tipo I ha gravità media ed è caratterizzata da uno sviluppo normale fino a quando un bambino ha 1 o 2 anni vecchio, a quel punto la crescita rallenta e si notano ritardi nello sviluppo. I sintomi non sono evidenti fino a 1 anno. L’aspettativa di vita per il tipo A è di circa 10-20 anni;
- la sindrome di Cockayne di tipo II, nota anche come “sindrome cerebro-oculo-facciale-scheletrica” o “sindrome di Pena-Shokeir di tipo B”, è il sottotipo più grave. I sintomi sono presenti alla nascita e il normale sviluppo cerebrale si interrompe dopo la nascita. La durata media della vita per i bambini di tipo B è fino a 7 anni;
- la sindrome di Cockayne di tipo III compare più tardi durante l’infanzia con sintomi più lievi rispetto agli altri tipi e una progressione più lenta del disturbo. Le persone con questo tipo di sindrome di Cockayne vivono fino all’età adulta, con una vita media di 40-50 anni.
Articoli sulle malattie progeroidi:
- Progeria (sindrome di Benjamin Button): cause, trasmissione, età di esordio e sintomi
- Progeria (sindrome di Benjamin Button): diagnosi, terapie e aspettativa di vita
- Sindrome di Werner (progeria degli adulti): cause, trasmissione, sintomi, diagnosi, cure, prognosi
- Xeroderma pigmentoso: cause, trasmissione, tipi, sintomi, diagnosi, terapia, prognosi e mortalità
- Malattia di DeSanctis-Cacchione: cause, sintomi, diagnosi e terapie
Leggi anche:
- Progeria: addio a Sam Berns, il bambino che invecchiava troppo in fretta
- Malattia di Charcot-Marie-Tooth: cause, sintomi, diagnosi, terapia
- Le malattie genetiche più diffuse al mondo
- Differenza tra autosomica dominante e recessiva con esempi
- Sindrome di Turner: cariotipo, cause, sintomi e segni caratteristici
- Sindrome di Klinefelter: cariotipo, cause, sintomi e cura
- Sindrome di Down: cause, sintomi in gravidanza e nei neonati
- Fibrosi cistica polmonare: cos’è, sintomi in neonati e bambini, cure
- Malattia di Huntington: cos’è, ereditarietà, come si trasmette, età di insorgenza
- Anemia falciforme: cosa significa, cause, sintomi e cure
- Differenze tra la distrofia muscolare di Duchenne e di Becker
- Talassemia: cos’è, sintomi, cure, differenti tipi ed alimentazione
- Celiachia: cos’è il glutine, in quali alimenti è contenuto ed in quali no?
- Sindrome di Noonan: cause, sintomi nel neonato, aspettative di vita
- Sindrome di Bloom: cause, sintomi, diagnosi e terapia
- Bimba nata con organi fuori dal corpo, glieli avvolgono con una pellicola
- Münchhausen per procura: perché questa mamma tortura il proprio bambino?
- Spina bifida e difetti di chiusura del tubo neurale nel feto: trasmissione, prevenzione, diagnosi e cura
- Mielomeningocele, spina bifida e schisi vertebrale: complicanze, cura, prevenzione
- Encefalocele: cause, sintomi, diagnosi, cura e prevenzione
- Cos’è un cromosoma ed a che serve?
- Cos’è un gene ed a che serve?
- Differenza tra malattia genetica, ereditaria e congenita
Dott. Emilio Alessio Loiacono
Medico Chirurgo
Direttore dello Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o unisciti al nostro gruppo Facebook o ancora seguici su Twitter, su Instagram, su Mastodon, su YouTube, su LinkedIn, su Tumblr e su Pinterest, grazie!
