
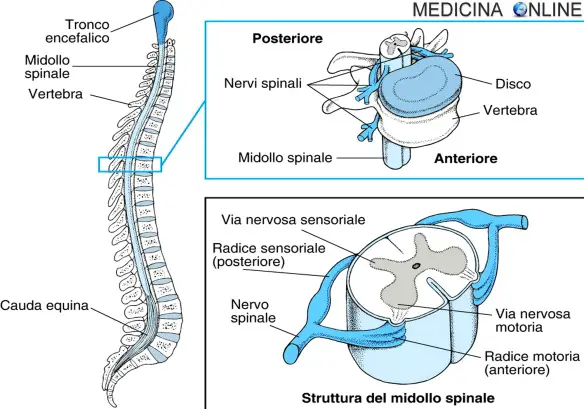
 Con “neuropatia periferica” si intende una patologia che colpisce il sistema nervoso periferico (SNP). La neuropatia può essere localizzata in un solo nervo (mononeuropatia), in più nervi con coinvolgimento simmetrico (polineuropatia) oppure in più nervi con coinvolgimento asimmetrico (mononeuropatia multipla). In questo articolo ci occuperemo brevemente della malattia di Charcot-Marie-Tooth.
Con “neuropatia periferica” si intende una patologia che colpisce il sistema nervoso periferico (SNP). La neuropatia può essere localizzata in un solo nervo (mononeuropatia), in più nervi con coinvolgimento simmetrico (polineuropatia) oppure in più nervi con coinvolgimento asimmetrico (mononeuropatia multipla). In questo articolo ci occuperemo brevemente della malattia di Charcot-Marie-Tooth.
Malattia di Charcot-Marie-Tooth
La malattia di Charcot-Marie-Tooth (spesso abbreviata con “CMT” o con “HMSN” da “Hereditary Motor and Sensory Neuropathy”), anche chiamata “atrofia muscolare peroneale, o neuropatia motorio-sensitiva ereditaria, è una rara sindrome neurologica ereditaria che interessa il sistema nervoso periferico e per tale motivo è inserita nell’ampio gruppo delle neuropatie periferiche).
Storia
La malattia di Charcot-Marie-Tooth deve il suo nome ai tre medici che per primi la descrissero: Jean-Martin Charcot, Pierre Marie, e Howard Henry Tooth.
Epidemiologia
Pur essendo rara, la malattia di Charcot-Marie-Tooth è comunque la malattia più diffusa tra le sindromi neurologiche ereditarie, con 35 casi ogni 100000 nascite.
Cause
Le cause sono ereditarie. A causa dei geni difettosi, la crescita della mielina dei nervi periferici risulta imperfetta e danneggiata causando danno all’assone.
Trasmissione
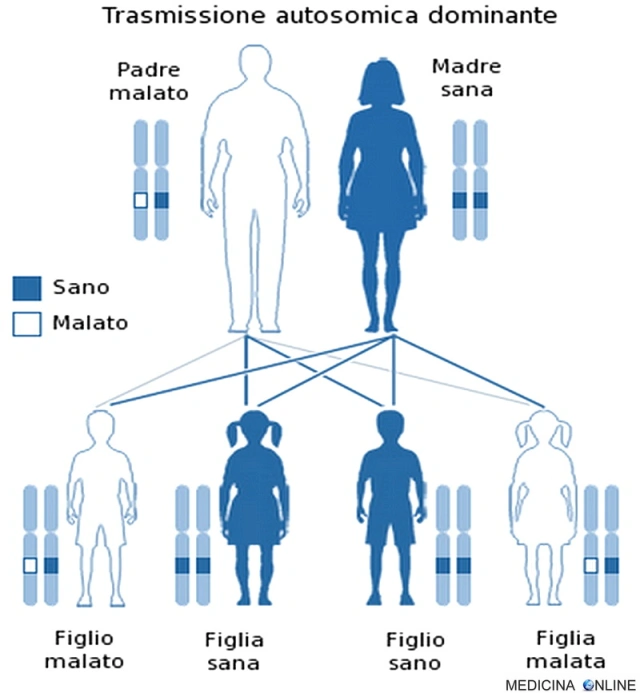
La malattia di Charcot-Marie-Tooth è trasmessa ai figli dai genitori nella maggior parte dei casi come carattere autosomico dominante. Una malattia è detta a trasmissione autosomica dominante quando basta una singola copia dell’allele difettoso per far sì che la malattia si esprima, a prescindere dal sesso (basta un solo genitore malato). Il figlio di un individuo affetto ha la probabilità del 50% di essere affetto, cioè 1 figlio su 2 è malato e può trasmettere a sua volta la malattia alla metà dei suoi figli. In questo caso non può esistere un “portatore sano” (cosa che invece si può verificare nella trasmissione autosomica recessiva): chi possiede l’allele alterato, ha la patologia, mentre chi non lo possiede è sano. Di conseguenza da due genitori sani nascono il 100% di figli sani, mentre se entrambi i genitori sono malati allora si avranno il 100% di figli malati.
Età di esordio
La malattia di Charcot-Marie-Tooth in genere si manifesta tra il primo e il secondo decennio di vita.
Sintomi e segni
L’esordio avviene nella tarda infanzia o nell’adolescenza con atrofia della muscolatura del piede e della gamba e successivamente delle mani e delle braccia. L’interessamento precoce dei muscoli peronieri e degli estensori delle dita determina lo sviluppo di un piede equinovaro. Sono compromesse sia la sensibilità profonda sia quella superficiale e i riflessi tendinei sono ridotti o assenti. La malattia ha un decorso estremamente lento, con lunghi periodi di stabilità. Raramente l’atrofia si estende prossimalmente al gomito e al terzo inferiore delle cosce. Le difficoltà nella deambulazione, che rappresentano il principale fattore di disabilità, sono dovute all’associazione di atassia e ipostenia. Il “piede cadente” (“foot drop”) e l’instabilità dell’articolazione dell’anca costituiscono segni addizionali che possono essere parzialmente corretti con interventi di artrodesi e con l’uso di stampelle.
Tipi
Esistono diversi tipi di malattia di Charcot-Marie-Tooth, con diversi sintomi e diversa gravità. Il tipo più diffuso è quello 1A, che determina perdita di tono muscolare e della sensibilità al tatto, in particolare agli arti inferiori, anche se le braccia possono essere interessate.
Diagnosi
La diagnosi si serve di anamnesi, esame obiettivo e vari test ed esami. Attualmente è disponibile un test genetico per le forme più comuni. L’elettromiografia è utile per la diagnosi: nella forma tipica (CMT1A) si osserva un rallentamento uniforme della velocità di conduzione in tutti i nervi, a differenza di ciò che si riscontra in qualsiasi altra polineuropatia acquisita. Esistono diverse varianti di questa patologia, in particolare una forma assonale (CMT2) e altre forme con ispessimento dei nervi; in alcune famiglie si associano anche segni di ipostenia e spasticità, oppure una patologia spinocerebellare.
Terapie
Non esiste una terapia specifica per la malattia di Charcot-Marie-Tooth, tuttavia la riabilitazione fornisce spesso buoni risultati. Una combinazione di accorgimenti nello stile di vita e/o l’assunzione attenta di farmaci possono contribuire a rallentare e ridurre gli effetti della sindrome. Per quel che riguarda i pazienti con deformità progressive, specie ai piedi, alle mani o alla colonna, la cura prevede fisioterapia, ortesi e chirurgia.
Prognosi e decorso
La malattia di Charcot-Marie-Tooth può portare ad esiti completamente differenti in base al tipo specifico: da insignificanti variazioni nelle capacità motorie all’atrofizzazione degli arti (che arrivano ad assumere una caratteristica forma assottigliata) con una serie di effetti correlati, da difficoltà di deambulazione e dolori muscolari fino (in rari casi) alla necessità permanente di sedia a rotelle. In genere non è una malattia letale, tuttavia in alcuni casi la muscolatura respiratoria viene danneggiata e può sopraggiungere la morte per insufficienza respiratoria.
Per concludere, vi lasciamo un link per l’associazione di pazienti affetti da CMT, www.acmt-rete.it
Leggi anche:
- Visita neurologica: svolgimento, esami, patologie, quando è necessaria?
- Sindrome di Guillain-Barré: cause, sintomi, diagnosi, cure, prognosi
- Differenza tra sistema nervoso centrale e periferico: anatomia e funzioni in sintesi
- Radicolopatia: significato, rimedi casalinghi, attività fisica, integratori, cure
- Elettromiografia: cos’è, quanto dura, costo, è dolorosa?
- Mielografia: cos’è, perché si esegue, come si esegue, quali sono i rischi?
- Neuropatia diabetica: sintomi e diagnosi di una complicanza del diabete
- Differenza tra tremori, spasmi, miotonia, crampi, fascicolazioni, tic
- Differenza tra discinesia, ipocinesia, ipercinesia, tardiva e primaria
- Fascicolazioni muscolari, il tremolio spontaneo di un muscolo: cause e cure
- Perdita della coordinazione muscolare: l’atassia
- Differenze tra aprassia, atassia, disprassia, afasia, disartria, anartria
- Differenza tra astenia, ipostenia, miastenia, ipotonia, nevrastenia, iperstenia, ipertonia
- Miastenia gravis e malattia del timo: cause, sintomi, diagnosi, terapia
- Sindrome miastenica di Lambert-Eaton e altre sindromi miasteniche
- Astenia, quando mancano le forze fisiche o mentali: cause, diagnosi, cure
- Nevrastenia (esaurimento nervoso): cause, diagnosi, cure
- Ipostenia (miastenia): cause, sintomi, diagnosi, terapie, complicanze, prognosi
- Ipotonia e atonia: definizione, etimologia, significato, esempi
- Ipotonia muscolare in neonati, adulti, anziani: cause, sintomi, cure, consigli
- Ipotonia nel lattante: cause, sintomi, diagnosi e trattamento
- Cos’è il tono muscolare, a che serve, come mantenerlo?
- Fibromialgia: sintomi, cause, cura e tender points
- Fibromialgia: dove si trovano i tender points che provocano dolore alla palpazione?
- Sindrome di Isaacs (neuromiotonia acquisita): cause, sintomi, cure
- Nervi cranici: anatomia, funzioni, schema, tabella, patologie
- Differenza tra nervo cranico, encefalico e spinale con esempi
- Puntura lombare: complicanze, risultati, è dolorosa, a che serve?
- Meningi: anatomia, funzioni e patologia in sintesi
- Liquido cefalorachidiano: dove si trova, perdita dal naso, prelievo
- Mielopatia: significato, tipi, sintomi, diagnosi,cure, consigli, prognosi
- Sclerosi multipla: cause, sintomi, diagnosi e prognosi
- Sclerosi laterale amiotrofica (SLA): cause, sintomi, diagnosi e prognosi
- Sindrome di Arnold-Chiari: linguaggio, aspettative di vita, invalidità, mortalità
- Siringomielia (malattia di Morvan): cause, sintomi, diagnosi, cure
- Siringobulbia: cause, sintomi, diagnosi e trattamento
- Test di Romberg: cos’è, a che serve, come si esegue
- Differenze tra sclerosi laterale amiotrofica e sclerosi multipla
- Atrofia muscolare progressiva: cause, sintomi, cura, aspettativa di vita
- Atrofia muscolare spinale: sintomi, trasmissione, tipi e cure
- Differenze tra atrofia muscolare progressiva e sclerosi laterale amiotrofica
- Malattia di Huntington: cos’è, ereditarietà, come si trasmette, età di insorgenza
- Iperstenia: definizione, significato, etimologia, fisiologica e patologica
- Ipertonia: definizione, significato, etimologia
- Ipertonia muscolare: cause, tipi, sintomi, diagnosi, terapie
- Distonia focale e generalizzata: tipi, sintomi, diagnosi e terapie
- Miotonia: cause, sintomi, diagnosi e terapie
- Demenza senile: cause, sintomi, decorso e cure
- Demenza da corpi di Lewy: cause, decorso, Parkinson, aspettativa di vita
- Differenza tra morbo di Alzheimer, demenza senile, vascolare e reversibile
- Mielite: infettiva, cervicale, dorsale, trasversa, si guarisce?
- Meningite: contagio, sintomi, vaccino, gravità e profilassi
- Segni meningei e irritazione meningi in bambini ed adulti
- Meningite: incubazione, fulminante, sintomi, contagio e cura
- Encefalite: conseguenze, è contagiosa, danni, si guarisce?
- Encefalite autoimmune da anticorpi anti-NMDA: trattamento, sintomi
- Meningismo: triade, segni, cause, diagnosi, definizione e cura
- Differenza tra meningite e meningismo: qual è più grave?
- Differenza tra meningite, encefalite, meningoencefalite, encefalomielite
- Differenza tra meningite virale e batterica
- Differenza tra virus e batteri: chi è più pericoloso? Diagnosi, sintomi e terapia
- Segni meningei e irritazione meningi in bambini ed adulti
- Tumore al cervello: cause, sintomi iniziali e tardivi, diagnosi, cura, sopravvivenza, aspettativa di vita
- Ipertensione endocranica: valori, cause, bradicardia, terapie
- Pseudotumor cerebri (ipertensione endocranica benigna) cause e cure
- Operato al cervello per un tumore mentre suona il clarinetto
- Tumore al cervello: operato mentre suona la chitarra e canta Yesterday
- Esoftalmo bi- e monolaterale: cause, gravità, conseguenze e cure
- Transilluminazione della testa di un neonato per la diagnosi di idrocefalo
- Idrocefalo: cause, terapia, conseguenze, aspettativa di vita
- Idrocefalo nel feto e neonatale: conseguenze e cura
- Pressione intracranica e pressione di perfusione cerebrale
- Sindrome della sella vuota: cause, sintomi, diagnosi e terapie
- Differenza idrocefalo iperteso, normoteso, comunicante, ostruttivo
- Macrocefalo in neonato e bambino: sintomi, cure e ritardo psicomotorio
- Shunt cerebrale e intervento per il drenaggio permanente
- Ventricoli cerebrali: anatomia e funzioni in sintesi
- Emorragia cerebrale da caduta e trauma cranico: sintomi, diagnosi e cure
- Emorragia cerebrale: non operabile, coma, morte, si può guarire?
- Emorragia cerebrale: operazione e tempi di riassorbimento
- Emorragia cerebrale: conseguenze, riabilitazione e recupero
- Emorragia subaracnoidea: cause, conseguenze, linee guida
- Ematoma subdurale: cos’è, da quale malattia è provocata, recidivo, decorso
- Coma da emorragia cerebrale: quanto può durare?
- Differenza tra proencefalo, mesencefalo, romboencefalo, telencefalo, diencefalo
- Differenza tra metencefalo e mielencefalo
- Emicrania con aura: cause, sintomi, diagnosi e trattamenti
- Emicrania senza aura: cause, sintomi, diagnosi e trattamenti
- Differenza tra emicrania con aura ed emicrania senza aura
- Cos’è l’aura emicranica?
- Ictus cerebrale emorragico e ischemico: cause, sintomi, diagnosi, cure, rischi
- Differenza tra ictus cerebrale ed attacco ischemico transitorio (TIA)
- Che cos’è un attacco ischemico transitorio (TIA)? Impara a riconoscerlo e potrai salvare una vita, anche la tua
- Aneurisma cerebrale rotto e non rotto: cause, sintomi, diagnosi e cura
- Cosa si prova e cosa succede quando si rompe un aneurisma cerebrale?
- Ictus, emorragia cerebrale cerebrale e TIA: cosa fare e cosa assolutamente NON fare
- Sindrome frontale o ipofrontalità: cause, sintomi, conseguenze, diagnosi, cure
- Malformazioni artero-venose cerebrali: sintomi e cura
- Ischemia: cos’è, cause, conseguenze, rischi, cure
- Necrosi: significato, definizione, sinonimo, cause, cure
- Trombo: cause, classificazione, trombosi venose, arteriose e sistemiche
- Infarto, ischemia, necrosi, aterosclerosi, trombo, embolo, ictus, miocardio… Facciamo chiarezza
- Tronco cerebrale (mesencefalo, ponte e bulbo) anatomia e funzioni in sintesi
- Morbo di Parkinson: cause, sintomi, decorso, terapie
- Morbo di Alzheimer: cause, sintomi, decorso, terapie
- Parestesie: significato, cause, rischi, diagnosi, cure, rimedi, esercizi
- Epilessia infantile ed in adulti: cause, sintomi, diagnosi, cosa fare
- Epilessia: come riconoscere un attacco e soccorrere un ammalato
- Differenza tra epilessia e convulsioni
- Differenza tra epilessia e sincope
- Differenza tra epilessia parziale e generalizzata
- Epilessia: riconoscere in tempo l’arrivo di una crisi e come comportarsi
- Epilessia infantile: come comportarsi col proprio figlio?
- Esame della sensibilità tattile, dolorifica, termica, vibratoria in neurologia
- Esame delle funzioni motorie e dei riflessi in neurologia
- Esame delle funzioni cerebrali superiori (corticali) in neurologia
- Asterissi (asterixis) in neurologia: caratteristiche, significato, esecuzione
- Torcicollo spasmodico e spasmi linguali, facciali, oromandibolari, della mano (distonie focali)
- Tic, ritmie, movimenti stereotipati, acatisia e trasalimento nel paziente neurologico
- Disturbi della stazione eretta e della deambulazione in neurologia
- Esame della motilità oculare e disturbi dei movimenti coniugati in neurologia
- Sordità, esame dell’udito e tecniche audiologiche speciali in neurologia
- Aprassia ideazionale, ideomotoria e melocinetica: sintomi, diagnosi, cure
- Aprassia: significato, etimologia, tipi, zone del cervello coinvolte
- Aprassia: test usati per la diagnosi e tipici errori aprassici
- Aprassia costruttiva (apractognosia): cause, sintomi, diagnosi, terapie
- Disartria: cause, sintomi, diagnosi, trattamento
- Non riconoscere i volti dei propri cari: la prosopagnosia, cause, test e cure
- Spina bifida e difetti di chiusura del tubo neurale nel feto: trasmissione, prevenzione, diagnosi e cura
- Mielomeningocele, spina bifida e schisi vertebrale: complicanze, cura, prevenzione
- Encefalocele: cause, sintomi, diagnosi, cura e prevenzione
- Anche gli esseri umani possono avere una coda
- Glasgow Coma Scale per la classificazione del coma
- Pediatric Glasgow Coma Scale in italiano: scala pediatrica del coma
- Differenza tra toracentesi, paracentesi e rachicentesi
- Narcolessia: cause, sintomi, cure e terapia farmacologica
- Catatonia: significato, definizione, cause, sinonimi e cure
- Cataplessia: causa, significato, nel sonno, cura ed etimologia
- Catalessia in medicina: cause, sintomi, nel sonno e cure
- Differenza tra catatonia, catalessia e cataplessia
- Paralisi del sonno e allucinazioni ipnagogiche: cause, pericoli, rimedi
- Morte cerebrale: diagnosi, sintomi, risveglio, durata, si può guarire?
- Coma: cause, risveglio, tipi, quanto dura, fasi, segni, irreversibile
- Differenza tra coma e coma farmacologico
- Elettroencefalogramma: preparazione, alterazioni, costo, rischi
- Sindrome locked-in: cause, riabilitazione, respirazione, cure
- Stato di minima coscienza: evoluzione, risveglio, riabilitazione
- Stato vegetativo: risveglio, riabilitazione, durata e caratteristiche
- Differenza tra stato vegetativo, di minima coscienza, coma, sonno e stato soporoso
- Differenza tra sindrome locked-in e stato vegetativo
- Differenza tra stato vegetativo persistente e permanente
- Differenza tra stato vegetativo e stato di minima coscienza
- Commozione cerebrale: cos’è, cosa fare, conseguenze, tempi di recupero
- Differenza tra commozione cerebrale, trauma cranico e contusione cerebrale
- Trauma cranico: ematoma, commotivo, sintomi tardivi, cosa fare
- Emorragia cerebrale: cause, sintomi premonitori, diagnosi e cura
- Differenza tra morte clinica, biologica, legale, apparente, improvvisa ed istantanea
- Poligono di Willis: anatomia e varianti anatomiche
- Emiplegia destra, sinistra, spastica, flaccida: significato e riabilitazione
- Emiparesi destra, sinistra, facciale e neonatale: cause, sintomi e cure
- Paraplegia: etimologia, significato, sintomi, cura e riabilitazione
- Tetraplegia: significato, cause, cure e riabilitazione
- Diplegia: definizione, cause e sintomi
- Differenza tra emiplegia, emiparesi, diplegia, paraplegia, tetraplegia
- Differenza emiparesi, diparesi, tetraparesi, monoparesi, triparesi
- Differenza tra paraplegia e diplegia
- Classificazione generale delle paresi e delle plegie
- Anosognosia e Sindrome neglect: significato, test e trattamento
- Sindrome neglect (negligenza spaziale unilaterale): cura e riabilitazione
- Sistema nervoso: com’è fatto, a che serve e come funziona
- Sistema nervoso simpatico: funzioni
- Sistema nervoso parasimpatico: funzioni
- Differenza tra afasia, disartria ed aprassia
- Area di Broca: funzioni ed afasia di Broca
- Area di Wernicke: funzioni ed afasia di Wernicke
- Differenza tra afasia di Broca e di Wernicke
- Cervelletto: anatomia esterna ed interna
- Cervelletto: le lesioni cerebellari più comuni
- Le funzioni del cervelletto: apprendimento e correzione dei movimenti del corpo
- Com’è fatto il cervello, a che serve e come funziona la memoria?
- Cervello maschile e femminile: quali sono le differenze?
- Sistema nervoso autonomo simpatico e parasimpatico: anatomia e funzioni
- Ipotalamo: anatomia, struttura e funzioni
- Differenze tra ipotalamo, ipofisi, neuroipofisi e adenoipofisi
- Differenza tra midollo osseo e spinale
- A cosa serve il midollo osseo?
- Differenza tra midollo osseo e cellule staminali
- Differenza tra midollo spinale e allungato
- Differenza tra epifisi, diafisi, metafisi ed ipofisi
- Patologie di ipotalamo e ipofisi
- Ipofisi (ghiandola pituitaria): anatomia, funzioni e ormoni secreti
- Asse ipotalamo-ipofisario: fisiologia e ormoni rilasciati
- Si può morire di epilessia?
- Sindrome pseudobulbare: cause, sintomi, diagnosi e terapie
- Malattia di Binswanger: cause, sintomi, diagnosi, cure
- Sistema piramidale e fascio genicolato: anatomia, decorso, funzioni
- Sistema extrapiramidale: anatomia, decorso, vie, funzioni, patologie
- Differenza tra via piramidale ed extrapiramidale
- Miclono: cause, sintomi, caratteristiche, quando preoccuparsi, cure
- Spasmi muscolari e mioclonie: da cosa sono causati?
- Spasmi muscolari e mioclonie: cause, diagnosi e cura delle contrazioni involontarie
- Spasmi muscolari e mioclonie: cura, trattamento e rimedi
- Spasmi muscolari e mioclonie: come si fa la diagnosi?
- Encefalopatia epatica: terapia, morte, sopravvivenza, dieta, prognosi
- Insufficienza epatica lieve, acuta e cronica: dieta e rischio di morte
- Cirrosi epatica e fegato: sintomi, dieta, diagnosi, terapia e prevenzione
- Funzionalità epatica; cos’è, cosa indica e come si misura
- Foetor hepaticus: causa, odore, definizione, significato
- Epatiti croniche: cosa sono, sintomi, diagnosi e cura
- Transaminasi alte, basse, cosa sono, cosa indicano e come si curano
- Ammoniemia (ammoniaca nel sangue): valori e dieta per iperammoniemia
- Encefalopatia uremica nell’uremia: sintomi e alito di urina
- Uremia: significato, valori, cause, sintomi, segni, morte
- Coma uremico e uremia: sintomi e morte del paziente
- Azotemia (Urea) alta o bassa: valori, cause, sintomi e cosa fare
- Insufficienza renale acuta: sintomi, terapia, linea guida, morte
- Insufficienza renale cronica: stadi, dieta, sintomi, diagnosi e terapia
- Emodialisi: come funziona, effetti collaterali e complicanze
- Esami per valutare funzionalità renale ed insufficienza renale
- Riflesso di Babinski positivo: sintomi, diagnosi, come evocarlo
- Segno di Babinski positivo nel neonato e nel bambino: che significa?
- Segno di Babinski nella sclerosi multipla e nella SLA
- Segno di Brudzinski positivo e negativo: semeiotica nella meningite
- Segno di Hoffman positivo in SLA e sclerosi multipla
- Segno di Kernig positivo e negativo: semeiotica nella meningite
- Segno di Lasègue positivo e negativo in semeiotica
- Segno di Wasserman (Lasègue inverso) positivo in semeiotica
- Segno di Binda: cos’è e come si esegue
- Segno di Amoss (o del tripode): cos’è e come si esegue
- Segno di Magnus-De Klein: cos’è e come si esegue
- Saturazione dell’ossigeno: valori normali e patologici in anziani e bambini
- Emogasanalisi arterioso: procedura, interpretazione, è dolorosa?
- Differenza tra ipossiemia e ipercapnia
- Differenza tra ipossiemia, ipossia, anossiemia ed anossia
- Ipossiemia: significato, valori, sintomi, conseguenze, rischi, cure
- Ipossia: valori, conseguenze, sintomi, cure
- Ipercapnia: valori, terapia, conseguenze e trattamento
- Anossia: definizione, cause, sintomi, sinonimo, cure
- Ipocapnia: significato, cause, valori, alcalosi respiratoria
- Ossigenoterapia: uso, controindicazioni, domiciliare, con maschera
- I migliori saturimetri professionali per uso ospedaliero e casalingo
- Differenza tra acidosi ed alcalosi, metabolica e respiratoria
- Alterazioni dell’equilibrio acido-base: acidosi ed alcalosi respiratorie e metabolica
- Differenza tra acidosi metabolica con gap anionico normale e aumentato
- Che significa malattia autoimmune? Spiegazione ed esempi
- Sistema piramidale e fascio genicolato: anatomia, decorso, funzioni
- Sistema extrapiramidale: anatomia, decorso, vie, funzioni, patologie
- Differenza tra via piramidale ed extrapiramidale
- Nervi degli arti superiori: anatomia e funzioni in sintesi
- Plesso brachiale: anatomia, funzioni e patologie in sintesi
- Plesso cervicale: anatomia, funzioni e patologie in sintesi
- Plesso lombare: anatomia, funzioni e patologie in sintesi
- Sindrome del tunnel carpale: prevenzione, diagnosi e cura di una dolorosa patologia
- Sindrome del tunnel radiale e sindrome del nervo interosseo posteriore
- Sindrome del tunnel cubitale: cause, sintomi, diagnosi e cura
- Sindrome del tunnel tarsale: cause, sintomi, diagnosi e terapia
- Nervi degli arti inferiori e plesso lombare: anatomia e funzioni
- Plesso sacrale: anatomia, rami e funzioni in sintesi
- Plesso coccigeo e nervo anococcigeo: anatomia e funzioni in sintesi
- Plesso pudendo e nervo pudendo: anatomia, rami e funzioni in sintesi
- Nervo sciatico (ischiatico): anatomia, funzioni e patologie
- Nervo femorale, muscolocutaneo, safeno, quadricipite: decorso e territorio di innervazione
- Nervo tibiale: anatomia, decorso e territorio di innervazione
- Nervo otturatorio: anatomia, decorso e territorio di innervazione
- Nervo ileoipogastrico: anatomia, decorso e territorio di innervazione
- Nervo ileoinguinale: anatomia, decorso e territorio di innervazione
- Nervo genitofemorale: anatomia, decorso e territorio di innervazione
- Nervo cutaneo laterale della coscia: anatomia, decorso e territorio di innervazione
- Nervo gluteo superiore e inferiore: anatomia, decorso e territorio di innervazione
- Nervo plantare laterale e mediale: anatomia, origine, decorso, rami
- Nervo peroneo comune: origine, percorso, innervazione motoria e sensitiva
- Nervi cranici: anatomia, funzioni, schema, tabella, patologie
- Gangli della base (nuclei della base): localizzazione, funzioni, patologie
- Globo pallido e pallido ventrale: anatomia e funzioni in sintesi
- Substantia nigra compatta e reticolata: anatomia e funzioni in sintesi
- Sistema dopaminergico: i circuiti nervosi della dopammina
- Talamo: anatomia, struttura, nuclei e funzioni in sintesi
- Ipotalamo: anatomia, struttura e funzioni
- Differenze tra ipotalamo, ipofisi, neuroipofisi e adenoipofisi
- Amigdala: connessioni, anatomia e funzioni in sintesi
- Ippocampo: anatomia, funzioni e ruolo nella memoria
- Locus ceruleus: anatomia, funzioni e connessioni in sintesi
Dott. Emilio Alessio Loiacono
Medico Chirurgo
Direttore dello Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o unisciti al nostro gruppo Facebook o ancora seguici su Twitter, su Instagram o su Pinterest, grazie!
