
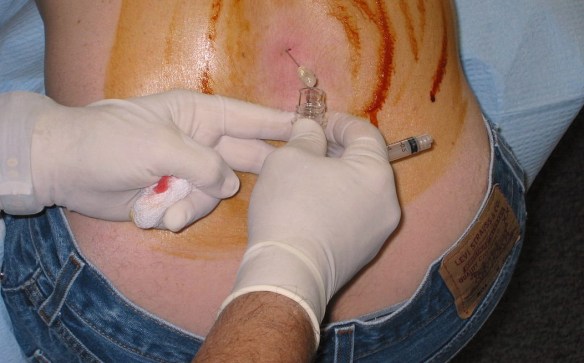
La rachicentesi è utile per la diagnosi
La sindrome di Guillain-Barré (chiamata anche “paralisi di Landry” o “sindrome di Guillain-Barré-Strohl“, o “poliradicoloneuropatia infiammatoria acuta demielinizzante” o “sindrome di Landry-Guillain-Barré“; in inglese “Guillain–Barré syndrome“, da cui l’acronimo “GBS“) è una radicolo-polinevrite acuta caratterizzata da paralisi progressiva agli arti con andamento disto-prossimale con probabile eziologia autoimmunitaria. La sindrome di Guillain-Barré appartiene al gruppo delle polineuropatie acute (anche denominate “poliradicolo-
neuropatie”) infiammatorie ed è potenzialmente mortale a causa dell’insufficienza respiratoria che è capace di determinare. La sindrome di Guillain-Barré, a differenza di sclerosi multipla e sclerosi laterale amiotrofica, è un disturbo del nervo periferico e generalmente non causa danni al cervello o al midollo spinale.
Storia
La sindrome di Guillain-Barré è stata identificata con precisione nel secondo decennio del ‘900 e prende il nome dai neurologi francesi Georges Guillain e Jean Alexandre Barré, e dal medico francese André Strohl. La prima descrizione della malattia fu compiuta dal medico francese Jean Landry molti anni prima: nel 1859.
Epidemiologia
La sindrome di Guillain-Barré ha carattere non epidemico e non stagionale ed interessa annualmente 1,5 soggetti ogni 100000 su scala mondiale. La sindrome di Guillain-Barré, pur essendo molto rara, è comunque la causa più comune di paralisi acuta non traumatica nel mondo. La sindrome interessa sia il sesso maschile e femminile, tuttavia gli uomini si ammalano 1,8 volte più delle donne. L’incidenza aumenta con l’età: vi è meno di un caso ogni 100000 persone sotto i 20 anni, circa 1 caso ogni 100000 persone sotto i 30 anni e circa 4 casi per 100000 persone oltre i 75 anni.
Cause
La causa della sindrome è sconosciuta, tuttavia è probabile che alla sua base vi sia un meccanismo autoimmune in cui il sistema immunitario, tramite “autoanticorpi”, attacca erroneamente materiale self dell’organismo, in particolare i nervi periferici, danneggiando il loro isolamento mielinico.
Fattori di rischio
Il probabile meccanismo autoimmunitario sembra essere scatenato da alcune patologie e condizioni, tra cui:
- infezioni;
- intervento chirurgico;
- vaccinazione (raro).
L’infezione sembra essere il fattore di rischio più importante: in circa due terzi dei casi l’ipostenia è preceduta, di 1-3 settimane, da un’infezione dell’apparato respiratorio o gastroenterico. Gli agenti più frequentemente isolati sono il virus di Epstein-Barr, il citomegalovirus, Mycoplasma pneumoniae e Campylobacter jejuni (enterite).
Sintomi e segni
I primi sintomi e segni sono:
- intorpidimento degli arti;
- parestesie;
- dolore muscolare;
- debolezza bilaterale delle gambe (“gambe gommose” o gambe che tendono a deformarsi con o senza disestesie);
- mancanza di forza.
La debolezza degli arti inferiori in genere precede quella degli arti superiori, inoltre tende a peggiorare con il tempo. Anche i muscoli del collo possono essere colpiti e metà dei pazienti sperimenta un coinvolgimento dei nervi cranici, che innervano la testa e il viso; ciò può portare alla debolezza dei muscoli del volto, a difficoltà di deglutizione, scialorrea, difficoltà nella respirazione e, talvolta, a debolezza dei muscoli oculari. La caratteristica principale della sindrome è l’ipostenia muscolare che evolve in modo più o meno simmetrico nell’arco di alcuni giorni fino a una o due settimane. Parestesie alle dita delle mani e dei piedi accompagnano, nella maggior parte dei casi, l’ipostenia. In genere viene interessata prima la muscolatura prossimale degli arti inferiori e successivamente quella del tronco, quella intercostale, delle braccia, del collo e dei muscoli del capo; a volte la progressione può avvenire in senso contrario. In tutti i casi i riflessi tendinei dapprima si riducono e poi scompaiono. Frequenti, ma solitamente di entità lieve e fugaci, sono i dolori muscolari e i segni e sintomi del sistema nervoso vegetativo.
Sindrome di Miller-Fisher
Una variante della sindrome di Guillain-Barré, che interessa i nervi cranici ed caratterizzata da atassia durante la deambulazione, areflessia e oftalmoplegia, prende il nome di sindrome di Miller-Fisher o “SMF”.
Diagnosi
La diagnosi della sindrome di Guillain-Barré si effettua principalmente in base all’anamnesi e all’esame obiettivo, che mostra rapido sviluppo della paralisi muscolare. aiutano nella diagnosi vari test diagnostici, in particolare la rachicentesi (puntura lombare), che permette l’analisi del liquor (liquido cefalorachidiano) e l’elettromiografia (EMG). Utile anche l’esame istologico dei nervi periferici.
- Nell’analisi del liquor si nota un aumento delle proteine liquorali.
- Nell’elettromiografia si nota la presenza di blocchi di conduzione e dispersione dei CMAP. I casi gravi provocano un danno assonale che si riflette nei segni EMG di denervazione; in casi estremi si rileva una completa ineccitabilità elettrica di alcuni nervi.
- All’esame istologico dei nervi periferici, ma soprattutto nelle fibre motorie e radici, si possono riscontrare infiltrati infiammatori costituiti da linfociti, monociti e macrofagi, questi ultimi probabilmente responsabili della demielinizzazione.
Diagnosi differenziale
La GBS rappresenta la polineuropatia a evoluzione più rapida ed è potenzialmente mortale (in pochi giorni progredisce verso una paralisi completa, con possibile morte per insufficienza respiratoria). Per questo motivo deve essere differenziata da altre forme di polineuropatia acuta (da difterite, porfiria, poliarterite, oppure tossica), nonché dalla miastenia gravis, dalla poliomielite e dalla mielite cervicale acuta. Tutti i dati disponibili indicano che si tratta di una patologia autoimmune, infiammatoria, demielinizzante. Raramente gli assoni rappresentano il bersaglio primario della reazione immunitaria.
Terapia
Per quanto concerne la terapia, il paziente dovrebbe essere ricoverato in un’unità di terapia intensiva. L’assistenza respiratoria deve essere instaurata ai primi segni di dispnea e di atelettasia (PO2 < 70 mmHg) o quando la capacità vitale è marcatamente ridotta <12-15 ml/kg). I pazienti che presentano una paralisi rapidamente progressiva e quelli che sono troppo deboli per camminare senza assistenza devono essere sottoposti a un ciclo di plasmaferesi (250 ml/kg di volume scambiato totale in 4-5 sedute) od a terapia con immunoglobuline per endovena (0,4 g/kg al giorno per 5 giorni). Entrambe queste terapie consentono di ridurre i tempi di guarigione. Studi controllati sull’uso degli steroidi non hanno dimostrato alcun beneficio.
Prognosi
La maggior parte dei pazienti va incontro a un recupero completo o pressoché completo (possono residuare lievi deficit motori agli arti inferiori), anche se la guarigione può richiedere da alcuni mesi a un anno o più. Circa il 3 – 7% dei pazienti non sopravvive, specie a causa dell’insufficienza respiratoria, ed in circa il 10% permane una grave disabilità residua. Durante la fase acuta circa il 15% dei pazienti sviluppa debolezza dei muscoli respiratori che richiedono il ricorso alla ventilazione meccanica. Circa il 3% dei pazienti incorre in uno o più episodi di ricaduta o può presentare una fluttuazione della sintomatologia per mesi o anni, come nella polineuropatia infiammatoria cronica demielinizzante.
Leggi anche:
- Visita neurologica: svolgimento, esami, patologie, quando è necessaria?
- Differenza tra sistema nervoso centrale e periferico: anatomia e funzioni in sintesi
- Che significa malattia autoimmune? Spiegazione ed esempi
- Radicolopatia: significato, rimedi casalinghi, attività fisica, integratori, cure
- Elettromiografia: cos’è, quanto dura, costo, è dolorosa?
- Mielografia: cos’è, perché si esegue, come si esegue, quali sono i rischi?
- Neuropatia diabetica: sintomi e diagnosi di una complicanza del diabete
- Differenza tra tremori, spasmi, miotonia, crampi, fascicolazioni, tic
- Differenza tra discinesia, ipocinesia, ipercinesia, tardiva e primaria
- Fascicolazioni muscolari, il tremolio spontaneo di un muscolo: cause e cure
- Perdita della coordinazione muscolare: l’atassia
- Differenze tra aprassia, atassia, disprassia, afasia, disartria, anartria
- Differenza tra astenia, ipostenia, miastenia, ipotonia, nevrastenia, iperstenia, ipertonia
- Miastenia gravis e malattia del timo: cause, sintomi, diagnosi, terapia
- Sindrome miastenica di Lambert-Eaton e altre sindromi miasteniche
- Astenia, quando mancano le forze fisiche o mentali: cause, diagnosi, cure
- Nevrastenia (esaurimento nervoso): cause, diagnosi, cure
- Ipostenia (miastenia): cause, sintomi, diagnosi, terapie, complicanze, prognosi
- Ipotonia e atonia: definizione, etimologia, significato, esempi
- Ipotonia muscolare in neonati, adulti, anziani: cause, sintomi, cure, consigli
- Ipotonia nel lattante: cause, sintomi, diagnosi e trattamento
- Cos’è il tono muscolare, a che serve, come mantenerlo?
- Fibromialgia: sintomi, cause, cura e tender points
- Fibromialgia: dove si trovano i tender points che provocano dolore alla palpazione?
- Sindrome di Isaacs (neuromiotonia acquisita): cause, sintomi, cure
- Differenza tra nervo cranico, encefalico e spinale con esempi
- Puntura lombare: complicanze, risultati, è dolorosa, a che serve?
- Meningi: anatomia, funzioni e patologia in sintesi
- Liquido cefalorachidiano: dove si trova, perdita dal naso, prelievo
- Mielopatia: significato, tipi, sintomi, diagnosi,cure, consigli, prognosi
- Sclerosi multipla: cause, sintomi, diagnosi e prognosi
- Sclerosi laterale amiotrofica (SLA): cause, sintomi, diagnosi e prognosi
- Sindrome di Arnold-Chiari: linguaggio, aspettative di vita, invalidità, mortalità
- Siringomielia (malattia di Morvan): cause, sintomi, diagnosi, cure
- Siringobulbia: cause, sintomi, diagnosi e trattamento
- Test di Romberg: cos’è, a che serve, come si esegue
- Differenze tra sclerosi laterale amiotrofica e sclerosi multipla
- Atrofia muscolare progressiva: cause, sintomi, cura, aspettativa di vita
- Atrofia muscolare spinale: sintomi, trasmissione, tipi e cure
- Differenze tra atrofia muscolare progressiva e sclerosi laterale amiotrofica
- Malattia di Huntington: cos’è, ereditarietà, come si trasmette, età di insorgenza
- Iperstenia: definizione, significato, etimologia, fisiologica e patologica
- Ipertonia: definizione, significato, etimologia
- Ipertonia muscolare: cause, tipi, sintomi, diagnosi, terapie
- Distonia focale e generalizzata: tipi, sintomi, diagnosi e terapie
- Miotonia: cause, sintomi, diagnosi e terapie
- Demenza senile: cause, sintomi, decorso e cure
- Demenza da corpi di Lewy: cause, decorso, Parkinson, aspettativa di vita
- Differenza tra morbo di Alzheimer, demenza senile, vascolare e reversibile
- Mielite: infettiva, cervicale, dorsale, trasversa, si guarisce?
- Meningite: contagio, sintomi, vaccino, gravità e profilassi
- Segni meningei e irritazione meningi in bambini ed adulti
- Meningite: incubazione, fulminante, sintomi, contagio e cura
- Encefalite: conseguenze, è contagiosa, danni, si guarisce?
- Encefalite autoimmune da anticorpi anti-NMDA: trattamento, sintomi
- Meningismo: triade, segni, cause, diagnosi, definizione e cura
- Differenza tra meningite e meningismo: qual è più grave?
- Differenza tra meningite, encefalite, meningoencefalite, encefalomielite
- Differenza tra meningite virale e batterica
- Differenza tra virus e batteri: chi è più pericoloso? Diagnosi, sintomi e terapia
- Segni meningei e irritazione meningi in bambini ed adulti
- Tumore al cervello: cause, sintomi iniziali e tardivi, diagnosi, cura, sopravvivenza, aspettativa di vita
- Ipertensione endocranica: valori, cause, bradicardia, terapie
- Pseudotumor cerebri (ipertensione endocranica benigna) cause e cure
- Operato al cervello per un tumore mentre suona il clarinetto
- Tumore al cervello: operato mentre suona la chitarra e canta Yesterday
- Esoftalmo bi- e monolaterale: cause, gravità, conseguenze e cure
- Transilluminazione della testa di un neonato per la diagnosi di idrocefalo
- Idrocefalo: cause, terapia, conseguenze, aspettativa di vita
- Idrocefalo nel feto e neonatale: conseguenze e cura
- Pressione intracranica e pressione di perfusione cerebrale
- Sindrome della sella vuota: cause, sintomi, diagnosi e terapie
- Differenza idrocefalo iperteso, normoteso, comunicante, ostruttivo
- Macrocefalo in neonato e bambino: sintomi, cure e ritardo psicomotorio
- Shunt cerebrale e intervento per il drenaggio permanente
- Ventricoli cerebrali: anatomia e funzioni in sintesi
- Emorragia cerebrale da caduta e trauma cranico: sintomi, diagnosi e cure
- Emorragia cerebrale: non operabile, coma, morte, si può guarire?
- Emorragia cerebrale: operazione e tempi di riassorbimento
- Emorragia cerebrale: conseguenze, riabilitazione e recupero
- Emorragia subaracnoidea: cause, conseguenze, linee guida
- Ematoma subdurale: cos’è, da quale malattia è provocata, recidivo, decorso
- Coma da emorragia cerebrale: quanto può durare?
- Differenza tra proencefalo, mesencefalo, romboencefalo, telencefalo, diencefalo
- Differenza tra metencefalo e mielencefalo
- Emicrania con aura: cause, sintomi, diagnosi e trattamenti
- Emicrania senza aura: cause, sintomi, diagnosi e trattamenti
- Differenza tra emicrania con aura ed emicrania senza aura
- Cos’è l’aura emicranica?
- Ictus cerebrale emorragico e ischemico: cause, sintomi, diagnosi, cure, rischi
- Differenza tra ictus cerebrale ed attacco ischemico transitorio (TIA)
- Che cos’è un attacco ischemico transitorio (TIA)? Impara a riconoscerlo e potrai salvare una vita, anche la tua
- Aneurisma cerebrale rotto e non rotto: cause, sintomi, diagnosi e cura
- Cosa si prova e cosa succede quando si rompe un aneurisma cerebrale?
- Ictus, emorragia cerebrale cerebrale e TIA: cosa fare e cosa assolutamente NON fare
- Sindrome frontale o ipofrontalità: cause, sintomi, conseguenze, diagnosi, cure
- Malformazioni artero-venose cerebrali: sintomi e cura
- Ischemia: cos’è, cause, conseguenze, rischi, cure
- Necrosi: significato, definizione, sinonimo, cause, cure
- Trombo: cause, classificazione, trombosi venose, arteriose e sistemiche
- Infarto, ischemia, necrosi, aterosclerosi, trombo, embolo, ictus, miocardio… Facciamo chiarezza
- Tronco cerebrale (mesencefalo, ponte e bulbo) anatomia e funzioni in sintesi
- Morbo di Parkinson: cause, sintomi, decorso, terapie
- Morbo di Alzheimer: cause, sintomi, decorso, terapie
- Parestesie: significato, cause, rischi, diagnosi, cure, rimedi, esercizi
- Epilessia infantile ed in adulti: cause, sintomi, diagnosi, cosa fare
- Epilessia: come riconoscere un attacco e soccorrere un ammalato
- Differenza tra epilessia e convulsioni
- Differenza tra epilessia e sincope
- Differenza tra epilessia parziale e generalizzata
- Epilessia: riconoscere in tempo l’arrivo di una crisi e come comportarsi
- Epilessia infantile: come comportarsi col proprio figlio?
- Esame della sensibilità tattile, dolorifica, termica, vibratoria in neurologia
- Esame delle funzioni motorie e dei riflessi in neurologia
- Esame delle funzioni cerebrali superiori (corticali) in neurologia
- Asterissi (asterixis) in neurologia: caratteristiche, significato, esecuzione
- Torcicollo spasmodico e spasmi linguali, facciali, oromandibolari, della mano (distonie focali)
- Tic, ritmie, movimenti stereotipati, acatisia e trasalimento nel paziente neurologico
- Disturbi della stazione eretta e della deambulazione in neurologia
- Esame della motilità oculare e disturbi dei movimenti coniugati in neurologia
- Sordità, esame dell’udito e tecniche audiologiche speciali in neurologia
- Aprassia ideazionale, ideomotoria e melocinetica: sintomi, diagnosi, cure
- Aprassia: significato, etimologia, tipi, zone del cervello coinvolte
- Aprassia: test usati per la diagnosi e tipici errori aprassici
- Aprassia costruttiva (apractognosia): cause, sintomi, diagnosi, terapie
- Disartria: cause, sintomi, diagnosi, trattamento
- Non riconoscere i volti dei propri cari: la prosopagnosia, cause, test e cure
- Spina bifida e difetti di chiusura del tubo neurale nel feto: trasmissione, prevenzione, diagnosi e cura
- Mielomeningocele, spina bifida e schisi vertebrale: complicanze, cura, prevenzione
- Encefalocele: cause, sintomi, diagnosi, cura e prevenzione
- Anche gli esseri umani possono avere una coda
- Glasgow Coma Scale per la classificazione del coma
- Pediatric Glasgow Coma Scale in italiano: scala pediatrica del coma
- Differenza tra toracentesi, paracentesi e rachicentesi
- Narcolessia: cause, sintomi, cure e terapia farmacologica
- Catatonia: significato, definizione, cause, sinonimi e cure
- Cataplessia: causa, significato, nel sonno, cura ed etimologia
- Catalessia in medicina: cause, sintomi, nel sonno e cure
- Differenza tra catatonia, catalessia e cataplessia
- Paralisi del sonno e allucinazioni ipnagogiche: cause, pericoli, rimedi
- Morte cerebrale: diagnosi, sintomi, risveglio, durata, si può guarire?
- Coma: cause, risveglio, tipi, quanto dura, fasi, segni, irreversibile
- Differenza tra coma e coma farmacologico
- Elettroencefalogramma: preparazione, alterazioni, costo, rischi
- Sindrome locked-in: cause, riabilitazione, respirazione, cure
- Stato di minima coscienza: evoluzione, risveglio, riabilitazione
- Stato vegetativo: risveglio, riabilitazione, durata e caratteristiche
- Differenza tra stato vegetativo, di minima coscienza, coma, sonno e stato soporoso
- Differenza tra sindrome locked-in e stato vegetativo
- Differenza tra stato vegetativo persistente e permanente
- Differenza tra stato vegetativo e stato di minima coscienza
- Commozione cerebrale: cos’è, cosa fare, conseguenze, tempi di recupero
- Differenza tra commozione cerebrale, trauma cranico e contusione cerebrale
- Trauma cranico: ematoma, commotivo, sintomi tardivi, cosa fare
- Emorragia cerebrale: cause, sintomi premonitori, diagnosi e cura
- Differenza tra morte clinica, biologica, legale, apparente, improvvisa ed istantanea
- Poligono di Willis: anatomia e varianti anatomiche
- Emiplegia destra, sinistra, spastica, flaccida: significato e riabilitazione
- Emiparesi destra, sinistra, facciale e neonatale: cause, sintomi e cure
- Paraplegia: etimologia, significato, sintomi, cura e riabilitazione
- Tetraplegia: significato, cause, cure e riabilitazione
- Diplegia: definizione, cause e sintomi
- Differenza tra emiplegia, emiparesi, diplegia, paraplegia, tetraplegia
- Differenza emiparesi, diparesi, tetraparesi, monoparesi, triparesi
- Differenza tra paraplegia e diplegia
- Classificazione generale delle paresi e delle plegie
- Anosognosia e Sindrome neglect: significato, test e trattamento
- Sindrome neglect (negligenza spaziale unilaterale): cura e riabilitazione
- Sistema nervoso: com’è fatto, a che serve e come funziona
- Sistema nervoso simpatico: funzioni
- Sistema nervoso parasimpatico: funzioni
- Differenza tra afasia, disartria ed aprassia
- Area di Broca: funzioni ed afasia di Broca
- Area di Wernicke: funzioni ed afasia di Wernicke
- Differenza tra afasia di Broca e di Wernicke
- Cervelletto: anatomia esterna ed interna
- Cervelletto: le lesioni cerebellari più comuni
- Le funzioni del cervelletto: apprendimento e correzione dei movimenti del corpo
- Com’è fatto il cervello, a che serve e come funziona la memoria?
- Cervello maschile e femminile: quali sono le differenze?
- Sistema nervoso autonomo simpatico e parasimpatico: anatomia e funzioni
- Ipotalamo: anatomia, struttura e funzioni
- Differenze tra ipotalamo, ipofisi, neuroipofisi e adenoipofisi
- Differenza tra midollo osseo e spinale
- A cosa serve il midollo osseo?
- Differenza tra midollo osseo e cellule staminali
- Differenza tra midollo spinale e allungato
- Differenza tra epifisi, diafisi, metafisi ed ipofisi
- Patologie di ipotalamo e ipofisi
- Ipofisi (ghiandola pituitaria): anatomia, funzioni e ormoni secreti
- Asse ipotalamo-ipofisario: fisiologia e ormoni rilasciati
- Si può morire di epilessia?
- Sindrome pseudobulbare: cause, sintomi, diagnosi e terapie
- Malattia di Binswanger: cause, sintomi, diagnosi, cure
- Sistema piramidale e fascio genicolato: anatomia, decorso, funzioni
- Sistema extrapiramidale: anatomia, decorso, vie, funzioni, patologie
- Differenza tra via piramidale ed extrapiramidale
- Miclono: cause, sintomi, caratteristiche, quando preoccuparsi, cure
- Spasmi muscolari e mioclonie: da cosa sono causati?
- Spasmi muscolari e mioclonie: cause, diagnosi e cura delle contrazioni involontarie
- Spasmi muscolari e mioclonie: cura, trattamento e rimedi
- Spasmi muscolari e mioclonie: come si fa la diagnosi?
- Encefalopatia epatica: terapia, morte, sopravvivenza, dieta, prognosi
- Insufficienza epatica lieve, acuta e cronica: dieta e rischio di morte
- Cirrosi epatica e fegato: sintomi, dieta, diagnosi, terapia e prevenzione
- Funzionalità epatica; cos’è, cosa indica e come si misura
- Foetor hepaticus: causa, odore, definizione, significato
- Epatiti croniche: cosa sono, sintomi, diagnosi e cura
- Transaminasi alte, basse, cosa sono, cosa indicano e come si curano
- Ammoniemia (ammoniaca nel sangue): valori e dieta per iperammoniemia
- Encefalopatia uremica nell’uremia: sintomi e alito di urina
- Uremia: significato, valori, cause, sintomi, segni, morte
- Coma uremico e uremia: sintomi e morte del paziente
- Azotemia (Urea) alta o bassa: valori, cause, sintomi e cosa fare
- Insufficienza renale acuta: sintomi, terapia, linea guida, morte
- Insufficienza renale cronica: stadi, dieta, sintomi, diagnosi e terapia
- Emodialisi: come funziona, effetti collaterali e complicanze
- Esami per valutare funzionalità renale ed insufficienza renale
- Riflesso di Babinski positivo: sintomi, diagnosi, come evocarlo
- Segno di Babinski positivo nel neonato e nel bambino: che significa?
- Segno di Babinski nella sclerosi multipla e nella SLA
- Segno di Brudzinski positivo e negativo: semeiotica nella meningite
- Segno di Hoffman positivo in SLA e sclerosi multipla
- Segno di Kernig positivo e negativo: semeiotica nella meningite
- Segno di Lasègue positivo e negativo in semeiotica
- Segno di Wasserman (Lasègue inverso) positivo in semeiotica
- Segno di Binda: cos’è e come si esegue
- Segno di Amoss (o del tripode): cos’è e come si esegue
- Segno di Magnus-De Klein: cos’è e come si esegue
- Saturazione dell’ossigeno: valori normali e patologici in anziani e bambini
- Emogasanalisi arterioso: procedura, interpretazione, è dolorosa?
- Differenza tra ipossiemia e ipercapnia
- Differenza tra ipossiemia, ipossia, anossiemia ed anossia
- Ipossiemia: significato, valori, sintomi, conseguenze, rischi, cure
- Ipossia: valori, conseguenze, sintomi, cure
- Ipercapnia: valori, terapia, conseguenze e trattamento
- Anossia: definizione, cause, sintomi, sinonimo, cure
- Ipocapnia: significato, cause, valori, alcalosi respiratoria
- Ossigenoterapia: uso, controindicazioni, domiciliare, con maschera
- I migliori saturimetri professionali per uso ospedaliero e casalingo
- Differenza tra acidosi ed alcalosi, metabolica e respiratoria
- Alterazioni dell’equilibrio acido-base: acidosi ed alcalosi respiratorie e metabolica
- Differenza tra acidosi metabolica con gap anionico normale e aumentato
Dott. Emilio Alessio Loiacono
Medico Chirurgo
Direttore dello Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o unisciti al nostro gruppo Facebook o ancora seguici su Twitter, su Instagram, su Mastodon, su YouTube, su LinkedIn, su Tumblr e su Pinterest, grazie!
