
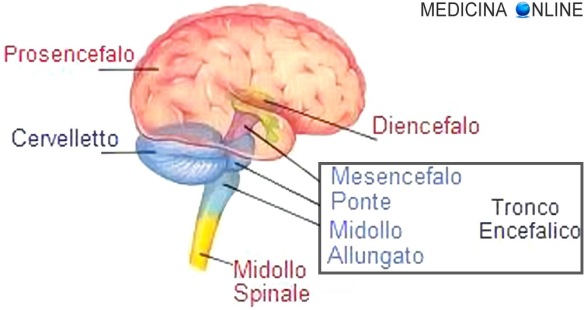
 L’encefalopatia necrotizzante acuta familiare (anche nota come “encefalopatia necrotizzante acuta ricorrente“, in inglese “familial acute necrotizing encephalopathy“) è una rarissima malattia neurologica potenzialmente fatale, caratterizzata da lesioni neuropatologiche che interessano in particolare il tronco cerebrale, il talamo e il putamen. Rircordo al lettore che l’encefalopatia necrotizzante acuta è un’encefalopatia rapidamente progressiva che si manifesta in individui, specie i bambini, altrimenti normali, associata a comuni infezioni virali, come l’influenza e la parainfluenza. La maggior parte dei casi è di forma sporadica e non ricorrente. Quasi tutti i casi conosciuti sono stati descritti in pazienti asiatici, sebbene siano stati segnalati casi isolati anche nei paesi occidentali. Solo recentemente, sono state scoperte mutazioni eterozigoti di un gene che codifica per una componente proteica del poro nucleare, chiamata Ran Binding Protein 2 (RANBP2) e tali mutazioni sono state riscontrate in un numero significativo di pazienti con la forma familiare o ricorrente di encefalopatia necrotizzante acuta, chiamata – appunto – encefalopatia necrotizzante acuta familiare o ricorrente.
L’encefalopatia necrotizzante acuta familiare (anche nota come “encefalopatia necrotizzante acuta ricorrente“, in inglese “familial acute necrotizing encephalopathy“) è una rarissima malattia neurologica potenzialmente fatale, caratterizzata da lesioni neuropatologiche che interessano in particolare il tronco cerebrale, il talamo e il putamen. Rircordo al lettore che l’encefalopatia necrotizzante acuta è un’encefalopatia rapidamente progressiva che si manifesta in individui, specie i bambini, altrimenti normali, associata a comuni infezioni virali, come l’influenza e la parainfluenza. La maggior parte dei casi è di forma sporadica e non ricorrente. Quasi tutti i casi conosciuti sono stati descritti in pazienti asiatici, sebbene siano stati segnalati casi isolati anche nei paesi occidentali. Solo recentemente, sono state scoperte mutazioni eterozigoti di un gene che codifica per una componente proteica del poro nucleare, chiamata Ran Binding Protein 2 (RANBP2) e tali mutazioni sono state riscontrate in un numero significativo di pazienti con la forma familiare o ricorrente di encefalopatia necrotizzante acuta, chiamata – appunto – encefalopatia necrotizzante acuta familiare o ricorrente.
Epidemiologia
La prevalenza dell’encefalopatia necrotizzante acuta familiare è minore a un caso per milione. La patologia è diffusa soprattutto nelle zone asiatiche del pianeta.
Esordio
L’esordio dell’encefalopatia necrotizzante acuta familiare avviene tipicamente nella prima infanzia. La patologia si presenta con episodi di encefalopatia acuta scatenati dalla febbre, solitamente tra gli 8 mesi e i 6 anni di età, sebbene in casi eccezionali sia stato descritto lo sviluppo di episodi di encefalopatia anche negli adulti. Le forme non familiari tipicamente insorgono dopo un episodio influenzale e possono interessare anche gli adulti.
Cause
L’encefalopatia necrotizzante acuta familiare è una malattia genetica a probabile trasmissione autosomica dominante con penetranza incompleta. È stata descritta in 11 soggetti appartenenti ad una stessa famiglia. La causa sembra essere correlata ad alterazione del gene RANBP2. Le mutazioni del gene RANBP2 non sono tuttavia l’unico allele che determina la suscettibilità allo sviluppo della forma familiare dell’encefalopatia necrotizzante acuta.
Sintomi e segni
Sintomi e segni dell’encefalopatia necrotizzante acuta familiare includono il vomito, le crisi epilettiche, la spasticità, la regressione del linguaggio, la rigidità e la postura anomala della testa. In alcuni pazienti persistono, dopo la fase acuta, gli esiti del deficit neurologico (debolezza muscolare, difficoltà di linguaggio, deficit cognitivo e disturbi comportamentali). La malattia è cronica in un caso ogni due.
Diagnosi
Il dubbio diagnostico dovbrebbe interessare il medico esperto già dall’anamnesi e dall’esame obiettivo. La diagnosi è generalmente fatta sulla base dei sintomi clinici, della storia familiare e dei risultati di test genetici e di laboratorio. La diagnostica per immagini è utile. Durante gli episodi acuti, la RM rileva iperintensità nel talamo e nel tronco encefalico sulle sequenze T2 e FLAIR. Insieme a questo, l’iperproteinorrachia e l’aumento delle aminotransferasi possono essere trovati come unici marcatori.
Decorso
Il decorso della malattia è generalmente rapido e può portare alla morte entro poche settimane o mesi dall’insorgenza dei sintomi.
Trattamento
Attualmente non esiste cura per l’encefalopatia necrotizzante acuta familiare.
Continua la lettura con:
- Encefalite emorragica necrotizzante acuta (leucoencefalite di Hurst)
- Neuromielite ottica (malattia di Devic o mielopatia necrotica)
- Malattie demielinizzanti: encefalomielite disseminata (post-infettiva)
- Sclerosi cerebrale diffusa (malattia di Schilder o encefalite periassiale diffusa)
- Sclerosi multipla: cause, sintomi, diagnosi e prognosi
- Guaina mielinica: anatomia, funzioni e patologie in sintesi
Leggi anche:
- Visita neurologica: svolgimento, esami, patologie, quando è necessaria?
- Sclerosi laterale amiotrofica (SLA): cause, sintomi, diagnosi e prognosi
- Test di Romberg: cos’è, a che serve, come si esegue
- Differenze tra sclerosi laterale amiotrofica e sclerosi multipla
- Atrofia muscolare progressiva: cause, sintomi, cura, aspettativa di vita
- Atrofia muscolare spinale: sintomi, trasmissione, tipi e cure
- Differenze tra atrofia muscolare progressiva e sclerosi laterale amiotrofica
- Riflesso di Babinski positivo: sintomi, diagnosi, come evocarlo
- Fascicolazioni muscolari, il tremolio spontaneo di un muscolo: cause e cure
- Differenza tra astenia, ipostenia, miastenia, ipotonia, nevrastenia, iperstenia, ipertonia
- Macrocefalo in neonato e bambino: sintomi, cure e ritardo psicomotorio
- Shunt cerebrale e intervento per il drenaggio permanente
- Tronco cerebrale (mesencefalo, ponte e bulbo) anatomia e funzioni in sintesi
- Morbo di Parkinson: cause, sintomi, decorso, terapie
- Morbo di Alzheimer: cause, sintomi, decorso, terapie
- Parestesie: significato, cause, rischi, diagnosi, cure, rimedi, esercizi
- Morte cerebrale: diagnosi, sintomi, risveglio, durata, si può guarire?
- Coma: cause, risveglio, tipi, quanto dura, fasi, segni, irreversibile
- Differenza tra coma e coma farmacologico
- Elettroencefalogramma: preparazione, alterazioni, costo, rischi
- Sindrome locked-in: cause, riabilitazione, respirazione, cure
- Stato di minima coscienza: evoluzione, risveglio, riabilitazione
- Stato vegetativo: risveglio, riabilitazione, durata e caratteristiche
- Poligono di Willis: anatomia e varianti anatomiche
- Emiplegia destra, sinistra, spastica, flaccida: significato e riabilitazione
- Emiparesi destra, sinistra, facciale e neonatale: cause, sintomi e cure
- Anosognosia e Sindrome neglect: significato, test e trattamento
- Sindrome neglect (negligenza spaziale unilaterale): cura e riabilitazione
Dott. Emilio Alessio Loiacono
Medico Chirurgo
Direttore dello Staff di Medicina OnLine
