
 La tirosinemia (in inglese “tyrosinemia” o “tyrosinaemia”) è malattia genetica metabolica congenita, cioè già presente alla nascita, causata da varie alterazioni genetiche trasmesse con modalità recessiva, in cui l’organismo non può metabolizzare efficacemente l’amminoacido tirosina, che tende ad accumularsi e determinare vari sintomi e segni.
La tirosinemia (in inglese “tyrosinemia” o “tyrosinaemia”) è malattia genetica metabolica congenita, cioè già presente alla nascita, causata da varie alterazioni genetiche trasmesse con modalità recessiva, in cui l’organismo non può metabolizzare efficacemente l’amminoacido tirosina, che tende ad accumularsi e determinare vari sintomi e segni.
Classificazione
La tirosinemia è essenzialmente di tre tipi:
- tirosinemia I o tirosinemia epatorenale;
- tirosinemia II o tirosinemia oculocutanea;
- tirosinemia III.
Cause
La tirosinemia ha cause genetiche. Tutti i tipi di tirosinemia derivano dalla disfunzione di vari geni nella via catabolica della fenilalanina e della tirosina e sono ereditate con modalità autosomica recessiva.
- La tirosinemia di tipo I deriva da una mutazione nel gene FAH, che codifica per l’enzima fumarilacetoacetasi. Come conseguenza del deficit di FAH, il substrato fumarilacetoacetato può accumularsi nelle cellule tubulari renali prossimali e negli epatociti, con conseguenti danni rispettivamente al rene e al fegato.
- La tirosinemia di tipo II deriva da una mutazione nel gene TAT, che codifica per l’enzima tirosina aminotransferasi. Come conseguenza del deficit di TAT, il substrato tirosina si accumula, causando anomalie oftalmologiche e dermatologiche.
- La tirosinemia di tipo III deriva da una mutazione nel gene HPD, che codifica per l’enzima 4-idrossifenilpiruvato diossigenasi. La tirosinemia di tipo III è la più rara delle tre condizioni, con solo pochi casi segnalati. La maggior parte di questi casi includeva disabilità intellettiva e disfunzione neurologica.
Trasmissione
Tutti e tre i tipi di tirosinemia sono malattie genetiche a trasmissione autosomica recessiva. Una malattia è detta a trasmissione autosomica recessiva quando l’allele alterato deve essere presente in coppia (omozigosi), cioè sono necessarie due copie dell’allele difettoso per far sì che la malattia si esprima, a prescindere dal sesso. Non basta un solo genitore portatore sano o malato, bensì entrambi i genitori devono essere portatori sani o malati. Il fenotipo quindi si esprime quando nel genotipo dell’individuo sono presenti entrambi gli alleli responsabili, fatto che spiega l’alta probabilità di sviluppare malattie genetiche in caso di incesto. Quindi:
- un individuo che possegga entrambi gli alleli alterati: è portatore ed è malato;
- un individuo che possegga solo un allele alterato: è portatore ma è sano;
- un individuo che non possegga nessun allele alterato: NON è portatore ed è sano.
Essere portatore sano vuol dire quindi NON avere la patologia ma possedere nel proprio genotipo un allele mutato, che può essere trasmesso alle generazioni successive.
Dalla combinazione delle possibili condizioni di genitori sani, malati e portatori sani, deriva la distribuzione probabilità che la malattia sia trasmessa ai figli:
- genitori malato-malato: la probabilità che il figlio/a nasca malato è del 100%;
- genitori sano-malato: la probabilità che il figlio/a nasca portatore sano è del 100%;
- genitori malato-portatore sano: la probabilità che il figlio/a nasca malato è del 50% e del 50% che nasca portatore sano;
- genitori sano-portatore sano: la probabilità che il figlio/a nasca sano è del 50% e del 50% che nasca portatore sano;
- genitori portatore-portatore: la probabilità che il figlio/a nasca portatore sano è del 50% mentre è del 25% che nasca sano o malato.
Se nessuno dei genitori ha un allele mutato, non c’è ovviamente alcuna trasmissione autosomica recessiva ed i figli saranno tutti sani e NON portatori dell’allele mutato.
Nell’immagine che segue, è raffigurata la tipica situazione in cui entrambi i genitori sono sani ma portatori dell’allele mutato:
- un figlio su quattro avrà entrambi gli alleli alterati e sarà malato ed ovviamente portatore;
- due figli su quattro avranno un allele normale ed uno alterato e saranno sani ma anche portatori;
- un figlio su quattro avrà entrambi gli alleli normali e sarà sano e NON portatore.

Le altre quattro situazioni possibili sono raffigurate nelle seguenti immagini:
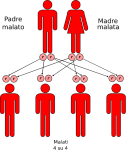
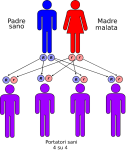
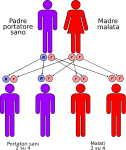
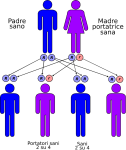
Sintomi e segni
I sintomi e segni della tirosinemia sono variabili in base al tipo (I, II o III). Parlando in generale, i sintomi e segni di una tirosinemia non trattata includono quelli relativi ad alterazione della funzionalità epatica e renale. E’ presente ritardo metale in alcuni casi. Sono presenti anomalie oftalmologiche e dermatologiche in altri casi. Senza trattamento, alcuni tipi di tirosinemia portano ad una insufficienza epatica che può essere fatale.
- la tirosinemia di tipo I o “tirosinemia epatorenale” si presenta soprattutto con alterazioni della funzionalità epatica, cui conseguono cirrosi e insufficienza, neuropatie periferiche, rachitismo e un caratteristico odore “di cavolo bollito”;
- la tirosinemia di tipo II o “tirosinemia oculocutanea” è caratterizzata da manifestazioni cliniche in particolar modo cutanee e oculari, con cheratosi del palmo della mano e della pianta dei piedi ed erosioni corneali associate a fotofobia;
- la tirosinemia di tipo III si manifesta con ipertirosinemia, a volte associata a ritardo mentale. La funzionalità epatica è normale.
Diagnosi
La tirosinemia di tipo I può essere rilevata tramite esami del sangue per la presenza di un metabolita fumarilacetoacetato, il succinilacetone, che è considerato un indicatore patognomonico della malattia.
La tirosinemia di tipo II può essere rilevata attraverso la presenza di livelli plasmatici di tirosina significativamente elevati e la diagnosi può essere confermata dal rilevamento di una mutazione in TAT nei fibroblasti in coltura.
La tirosinemia di tipo III può essere diagnosticata rilevando una mutazione in HPD nei fibroblasti in coltura.
Leggi anche: Insufficienza renale cronica: stadi, dieta, sintomi, diagnosi e terapia
Trattamento
Il trattamento varia a seconda del tipo specifico di tirosinemia. Nella maggior parte dei casi è necessaria una dieta a basso contenuto proteico combinata con l’uso di una formula appositamente progettata per fornire proteine. L’esperienza con il nitisinone ha dimostrato che è efficace, soprattutto se iniziato entro il primo mese di vita, ed è ora il corso standard di trattamento. È un inibitore della 4-idrossifenilpiruvato diossigenasi indicato per il trattamento della tirosinemia ereditaria di tipo 1 (HT-1) in combinazione con la restrizione dietetica di tirosina e fenilalanina. Il trapianto di fegato è indicato per i pazienti con tirosinemia di tipo I che non rispondono al nitisinone, così come per quelli con insufficienza epatica acuta ed epatomi.
Prognosi
Oggi, la tirosinemia viene sempre più rilevata nei test di screening neonatale prima che compaiano i sintomi. Con una gestione precoce e per tutta la vita che comporta una dieta a basso contenuto proteico, una formula proteica speciale e talvolta farmaci, le persone con tirosinemia si sviluppano normalmente, sono sane e vivono una vita normale.
Leggi anche:
- Fenilchetonuria: sintomi, valori, alimentazione, diagnosi
- Alcaptonuria: cause, trasmissione, sintomi, diagnosi e terapie
- Ocronosi endogena ed esogena: cause, trasmissione, sintomi, diagnosi e terapie
- Mioglobinuria genetica ricorrente: cause, sintomi, diagnosi
- Mioglobinuria (mioglobina nelle urine): significato, cause, esame delle urine, cure
- Esami per valutare funzionalità renale ed insufficienza renale
- Esame delle urine completo con urinocoltura: come farlo e capire i risultati
- Emoglobina nelle urine (emoglobinuria): cause, sintomi e terapia
- Ematuria da sforzo e da esercizio fisico: cause e terapia
- Proteinuria 24 ore alta: cause, tipi, valori e terapie
- Batteriuria: asintomatica, non significativa, valori normali, cura
- Scura o chiara, liquida o schiumosa: la tua urina rivela la tua salute
- Iperuricemia, ipericosuria, acido urico nel sangue e nelle urine
- Differenza tra emoglobina e mioglobina
- Differenza tra ematuria iniziale, terminale e totale ed ipotesi diagnostiche
- Emodialisi: come funziona, effetti collaterali e complicanze
- Quali sono i sintomi di malattia o cattivo funzionamento dei reni?
- Differenza tra esame delle urine ed urinocoltura
- Mi alzo spesso di notte per urinare: quali sono le cause e le cure?
- CPK (CK) alto o basso: cause, conseguenze, valori, cure, cosa indica
- Miastenia gravis e malattia del timo: cause, sintomi, diagnosi, terapia
- Le tue feci dicono se sei in salute: con la Scala di Bristol impara ad interpretarle
- Emoglobina bassa, alta, cause e valori normali
- Differenza tra sindrome nefritica e nefrosica
- Differenza tra proteinuria transitoria, persistente ed ortostatica
- Uroflussometria: indicazioni, preparazione, come si esegue
- Vescica: dove si trova, anatomia, funzioni e patologie frequenti in sintesi
- Rene: anatomia, funzioni e patologie in sintesi
- Azotemia (Urea) alta o bassa: valori, cause, sintomi e cosa fare
- Tumore alla vescica: terapie, asportazione, si può guarire?
- Bruciore e stimoli frequenti di urinare: cistite, sintomi e cure
- Perché viene la cistite e come curarla?
- Differenza tra anuria e ritenzione urinaria
- Esame delle urine completo con urinocoltura: come fare e capire i risultati
- Urodinamica: cos’è, a che serve e come funziona
- Minzione: come funziona l’emissione di urina e come si controlla
- Uretra maschile e femminile: anatomia, funzioni e patologie in sintesi
- Uretere: dove si trova, anatomia, funzioni e patologie in sintesi
- Fa male trattenere l’urina troppo a lungo? Per quale motivo?
- Differenze tra apparato urinario maschile e femminile
- Quante volte al giorno è normale urinare? Vescica iperattiva e ansia
- Prostata: anatomia, dimensioni, posizione e funzioni in sintesi
- Ecografia prostatica transrettale: come si svolge, è dolorosa, a che serve?
- PSA totale e free alto: capire i risultati dell’esame e rischio di tumore alla prostata
- Esplorazione rettale digitale della prostata: fa male? A che serve?
- Prostata ingrossata ed infiammata: ecco cosa fare per mantenerla in salute
- Vescica neurogena disinibita, riflessa, autonoma, atonica
- Albumina ed albuminemia alta o bassa: cause, valori e terapie
- Parassiti e vermi nelle feci: sintomi e come eliminarli con farmaci e rimedi naturali
- Coprocoltura e antibiogramma: procedura e perché si eseguono
- Esame e raccolta delle feci: come si fa nel modo corretto ed a che serve
- Differenza tra globuli rossi, bianchi e piastrine
- Feci con sangue, muco, cibo: quando preoccuparsi?
- Emocromo: guida completa a tutti i valori del sangue normali e patologici
- Emocromo: valori di riferimento e significato clinico [SCHEMA]
- Ematocrito (HCT): basso, alto, in gravidanza, valori normali e interpretazione
- Indici corpuscolari MCV, MCH, MCHC, RDW: cosa sono ed a che servono
- Volume corpuscolare medio (MCV): alto, basso, valori normali e significato
- MCH alto, basso, valori normali ed interpretazione
- MCHC alto, basso, valori normali ed interpretazione
- RDW alto, basso, valori normali ed interpretazione
- Ormoni tiroidei: differenza T3 e T4, valori normali e patologici
- TSH alto, basso e valori normali: qual è il significato clinico?
- Tireoglobulina alta, bassa, valori normali ed interpretazione
- Globuli rossi (eritrociti) alti, bassi, valori normali e interpretazione
- Globuli bianchi (leucociti) alti, bassi, valori normali ed interpretazione
- Eosinofili alti, bassi, valori normali ed interpretazione
- Neutrofili alti, bassi, valori normali ed interpretazione
- Basofili alti, bassi, valori normali ed interpretazione
- Differenza tra anemia ed anemia mediterranea (talassemia)
- Differenza tra anemia e leucemia
- Perché la mononucleosi è chiamata anche “malattia del bacio”?
- Leucemia: sintomi, cause, cure e le diverse forme
- Leucemia mieloide acuta: cause, sintomi, diagnosi e cura
- Mieloma multiplo: cause, sintomi, diagnosi e cura
- Microcitemia (talassemia) : cause, sintomi, diagnosi e cura
- Differenza tra anemia e microcitemia
- Differenza tra anemia megaloblastica e perniciosa
- Differenza tra anemia mediterranea e falciforme
- Differenza tra emoglobina fetale ed adulta
- Differenza tra emoglobina, ferro, ferritina e transferrina
- Differenza tra emoglobina e globuli rossi
- Differenza tra emocromo ed ematocrito
- Surrene: anatomia, funzioni e patologie in sintesi
- Differenza tra renella e calcoli renali
- Rene: anatomia, funzioni e patologie in sintesi
- Differenza tra surrene e rene
- Differenza tra rene policistico e multicistico
- Differenza tra rene destro e sinistro
- Differenza tra nefrologo ed urologo: patologie e competenze specifiche e comuni
- Testosterone basso, alto, valori normali ed interpretazione
- Ormone follicolo stimolante (FSH) alto, basso, valori normali e significato
- Lingua bianca, impastata, spaccata: cause e quando è pericolosa
- Lingua bianca, impastata, spaccata: cure e rimedi naturali
- Smegma femminile, vulviti e cattivi odori vaginali: cura e prevenzione
- Mi dicevano “sei grassa” così decisi che non avrei mangiato più. Mai più. La testimonianza di una paziente anoressica
- Anoressia: le immagini drammatiche di un corpo che non esiste più
- Si mette a dieta e perde 60 kg. La ragione per cui lo fa vi lascerà senza parole
- Apparato urinario: anatomia e fisiologia [SCHEMA]
- Filtrazione glomerulare, riassorbimento e secrezione
- Differenza tra arteriola afferente ed efferente: struttura e funzioni
- Glomerulo renale: schema, funzione e flusso ematico renale
- Qual è la differenza tra arteria e vena?
- Trombo: cause, classificazione, trombosi venose, arteriose e sistemiche
- Differenza tra trombosi arteriosa e venosa profonda e superficiale
- Differenza tra trombo e placca aterosclerotica
- Differenza tra pressione arteriosa e venosa
- Differenza tra pressione massima (sistolica), minima (diastolica) e differenziale
- Pressione arteriosa: valori normali e patologici
- Pressione alta (ipertensione arteriosa): sintomi, cause, valori e cure
- Perché la pressione arteriosa alta (ipertensione) è pericolosa?
- Ipertermia maligna: cause, fattori di rischio, sintomi, rischi, cure, mortalità
- Rabdomiolisi: sintomi, da statine, conseguenze, valori
- Ustione di terzo grado: cosa fare e tempi di guarigione della pelle
Dott. Emilio Alessio Loiacono
Medico Chirurgo
Direttore dello Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o unisciti al nostro gruppo Facebook o ancora seguici su Twitter, su Instagram, su Tumblr e su Pinterest, grazie!
