
 Con “fenilchetonuria” o “iperfenilalaninemia di tipo I” in medicina si indica la presenza di elevate concentrazione di fenilpiruvato nelle urine e dell’aminoacido fenilalanina nel sangue.
Con “fenilchetonuria” o “iperfenilalaninemia di tipo I” in medicina si indica la presenza di elevate concentrazione di fenilpiruvato nelle urine e dell’aminoacido fenilalanina nel sangue.
Nell’uso comune, con “fenilchetonuria” ci si riferisce più spesso alla “sindrome fenilchetonurica” (abbreviata con “PKU“), la più comune malattia pediatrica genetica esistente, dovuta a diversi tipi di mutazioni recessive di un gene localizzato sul cromosoma 12 (che codifica per la fenilalanina idrossilasi), tutte accomunate dal fatto di produrre come effetto finale fenilchetonuria e iperfenilalaninemia, cioè le prima citate condizioni di eccesso di fenilpiruvato nelle urine e di fenilalanina nel sangue. La forma più comune è nota come fenilchetonuria classica, caratterizzata da una sintomatologia più grave. Sono state inoltre descritte altre forme più lievi:
- forma lieve (PKU lieve);
- iperfenilalaninemia lieve (HPA lieve o HPA- non PKU).
Diffusione
La fenilchetonuria è classificata come malattia rara, avendo una frequenza di 1-5 bimbi malati ogni 10.000 nati vivi. La prevalenza è molto alta in Turchia dove è di 1 malato su 4.000 nati vivi, mentre è molto bassa nei popoli finlandesi, africani e giapponesi.
Cause e patogenesi
La fenilchetonuria o iperfenilalaninemia di tipo I è una malattia autosomica recessiva dovuta a mutazioni nel gene PAH (Phenylalanine Hydroxylase) codificante per l’enzima fenilalanina idrossilasi, attivo in particolare nel fegato. Sono note circa 500 mutazioni differenti a carico di questo gene. La fenilalanina idrossilasi è un enzima che, con la collaborazione della tetraidrobiopterina (BH4) come cofattore, trasforma l’amminoacido essenziale fenilalanina in tirosina. Quando l’enzima fenilalanina idrossilasi non è funzionante, la fenilalanina si accumula nel sangue e in alcuni tessuti del corpo (oltre 20 mg/dL mentre i livelli normali sono di 1 mg/dL). La fenilalalina accumulata viene in parte convertita in fenilpiruvato e fenilacetato, i quali vengono escreti attraverso le urine. La carenza di tirosina, dovuta all’inefficiente conversione di fenilalanina in tirosina, porta a bassi livelli di tirosina e conseguentemente a un’insufficiente produzione di molecole da essa derivate, come i neurotrasmettitori adrenalina, noradrenalina, dopamina e il precursore della melanina, la DOPA. Elevate concentrazioni di fenilalanina nel cervello possono provocare ritardo mentale, ritardo nell’accrescimento e morte precoce. Non è ancora noto il meccanismo con il quale la fenilalanina provochi ritardo mentale, ma si pensa che saturi i trasportatori di alcuni amminoacidi presso la barriera ematoencefalica determinando da una parte una concentrazione troppo elevata di questa sostanza nel cervello e d’altra parte una diminuzione dei livelli di altri amminoacidi con cui compete a livello del trasportatore. Inoltre, dal momento che i livelli di tirosina risultano bassi e che la fenilalanina è un inibitore di enzimi come la triptofano idrossilasi, c’è una minore produzione di molti neurotrasmettitori fondamentali nei circuiti nervosi, cioè dopamina, serotonina, adrenalina e noradrenalina. Sembra che elevate concentrazioni di fenilalanina rallentino anche lo sviluppo neuronale determinando neuroni più piccoli della media che hanno più difficoltà nel formare sinapsi con altri neuroni. Alla fenilchetonuria si affiancano patologie note come iperfenilalaninemie (di tipo II e III) dovute o ad un’attività ridotta della fenilalanina idrossilasi oppure per livelli troppo bassi di tetraidrobiopterina dovuti a disfunzioni degli enzimi diidropteridina reduttasi o 6-piruvoiltetraidropterina sintetasi.
Trasmissione
La fenilchetonuria è una malattia autosomica recessiva, il che significa che si verifica quando l’allele alterato deve essere presente in coppia (omozigosi), cioè sono necessarie due copie dell’allele difettoso per far sì che la malattia si esprima, a prescindere dal sesso. Non basta un solo genitore portatore sano o malato, bensì entrambi i genitori devono essere portatori sani o malati. Il fenotipo quindi si esprime quando nel genotipo dell’individuo sono presenti entrambi gli alleli responsabili, fatto che spiega l’alta probabilità di sviluppare malattie genetiche in caso di incesto. Quindi:
- un individuo che possegga entrambi gli alleli alterati: è portatore ed è malato;
- un individuo che possegga solo un allele alterato: è portatore ma è sano;
- un individuo che non possegga nessun allele alterato: NON è portatore ed è sano.
Essere portatore sano vuol dire quindi NON avere la patologia ma possedere nel proprio genotipo un allele mutato, che può essere trasmesso alle generazioni successive.
Probabilità di trasmissione ai figli
Dalla combinazione delle possibili condizioni di genitori sani, malati e portatori sani, deriva la distribuzione probabilità che la malattia sia trasmessa ai figli:
- genitori malato-malato: la probabilità che il figlio/a nasca malato è del 100%;
- genitori sano-malato: la probabilità che il figlio/a nasca portatore sano è del 100%;
- genitori malato-portatore sano: la probabilità che il figlio/a nasca malato è del 50% e del 50% che nasca portatore sano;
- genitori sano-portatore sano: la probabilità che il figlio/a nasca sano è del 50% e del 50% che nasca portatore sano;
- genitori portatore-portatore: la probabilità che il figlio/a nasca portatore sano è del 50% mentre è del 25% che nasca sano o malato.
Se nessuno dei genitori ha un allele mutato, non c’è ovviamente alcuna trasmissione autosomica recessiva ed i figli saranno tutti sani e NON portatori dell’allele mutato.
Nell’immagine che segue, è raffigurata la tipica situazione in cui entrambi i genitori sono sani ma portatori dell’allele mutato:
- un figlio su quattro avrà entrambi gli alleli alterati e sarà malato ed ovviamente portatore;
- due figli su quattro avranno un allele normale ed uno alterato e saranno sani ma anche portatori;
- un figlio su quattro avrà entrambi gli alleli normali e sarà sano e NON portatore.

Le altre quattro situazioni possibili sono raffigurate nelle seguenti immagini:
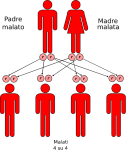
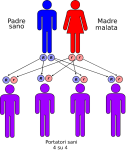
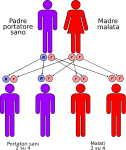
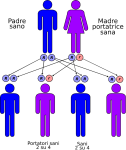
IMPORTANTE: La consulenza genetica è sempre consigliata ai familiari dei pazienti affetti.
Sintomi iniziali
I bambini affetti da fenilchetonuria generalmente non dimostrano sintomi particolari alla nascita ma successivamente (spesso prima dei due mesi di vita) tendono a sviluppare alcuni segni e sintomi, tra cui:
- malessere generale;
- rash cutaneo;
- nausea;
- vomito;
- irritabilità;
- eczema;
- tremori;
- ritardo dello sviluppo;
- deficit di accrescimento;
- microcefalia;
- epilessia;
- odore particolare “di muffa”;
- ipertono muscolare;
- iperreflessia tendinea.
La scarsa presenza di tirosina non permette che sia convertita in quantità sufficienti di DOPA e quindi di melanina, conseguentemente si verifica depigmentazione col risultato che i bimbi affetti da fenilchetonuria hanno spesso:
- carnagione chiara,
- occhi azzurri o verdi;
- capelli biondi.
Sintomi tardivi
Il sintomo principale a lungo termine è il ritardo mentale progressivo, che si associa ai sintomi e segni già elencati precedentemente. La fenilchetonuria classica ha sintomi e segni più gravi rispetto alla PKU lieve o alla HPA lieve.
Diagnosi
Viste le gravi conseguenze a cui vanno incontro i fenilchetonurici, gli stati europei e americani hanno istituito indagini su tutti i neonati per la diagnosi precoce della PKU. Le analisi vengono effettuate mediante la spettrometria di massa tandem. In uso anche il test di Guthrie che consiste nel porre una goccia di sangue su un disco di carta da filtro, messo in seguito in contatto con una coltura del batterio Bacillus subtilis a cui è aggiunta β-2-tienialanina, che inibisce la crescita batterica. In presenza di fenilalanina, l’inibizione viene contrastata e i batteri sopravvivono. Una continuazione della crescita batterica è quindi segnale di alti livelli di fenilalanina, ed è necessario che il neonato sia sottoposto a ulteriori analisi. Oppure, la diagnosi viene raggiunta utilizzando il sangue prelevato alla nascita (almeno dopo 48 ore) e analizzandolo con un cromatografo per amminoacidi. Con questo test si hanno meno falsi positivi.
Terapia e profilassi
La terapia principale per la fenilchetonuria è una dieta con un apporto di fenilalanina estremamente limitato e controllato: la fenilalanina è un aminoacido e si trova soprattutto nei cibi che contengono proteine, ma – dal momento che è amminoacido essenziale (quindi non sostituibile con altre molecole) ed indispensabile alla produzione delle proteine dei tessuti dell’organismo – l’apporto di fenilalanina NON deve essere azzerato del tutto, bensì limitato, in modo che i valori di fenilalanina nel sangue rimangano entro limiti accettabili. Il fabbisogno giornaliero di fenilalanina è quindi stimato in seguito ai controlli ematici periodici.
Valori normali e patologici di fenilalanina
I valori ottimali di fenilalanina ematica oscillano tra i 2 e 6 mg/100 ml di sangue (120 – 360 micromol/L) : valori superiori o inferiori a tale intervallo di valori, sono patologici ed indicano rispettivamente un apporto eccessivo o insufficiente di fenilalanina con la dieta.
Dieta consigliata
Nell’alimentazione dei pazienti con fenilchetonuria esistono alcuni alimenti permessi, altri consumabili ma con moderazione, altri sconsigliati e da evitare.
Alimenti permessi:
- la maggior parte della frutta e verdura di stagione;
- vari tipi di olio;
- cibi ipoproteici;
- burro;
- margarina;
- succhi di frutta;
- fecola di mais;
- tè;
- caffè;
- caramelle;
- zucchero;
- miele;
- marmellata;
- spezie;
- sale.
Leggi anche: Dieta ipoproteica ed aproteica: cosa mangiare e chi la deve seguire
Alimenti da consumare con moderazione:
- soia;
- avocado;
- panna;
- banana;
- cereali;
- riso;
- mais;
- patate;
- piselli;
- broccoli;
- cavoletti di Bruxelles;
- asparagi;
- spinaci.
Alimenti sconsigliati, da assumere con ESTREMA moderazione o da evitare:
- carne;
- pesce;
- uova;
- molluschi;
- crostacei;
- noci;
- birra;
- farine;
- pane;
- pasta;
- biscotti;
- legumi secchi;
- yogurt;
- formaggio;
- frutta secca;
- latte;
- cioccolato.
ATTENZIONE: anche consumare un alimento permesso dalla dieta può essere un errore, se ne mangiate quantità eccessive. MAI esagerare! La dieta deve essere stilata solo dal medico. In ogni caso chiedete SEMPRE consiglio al medico prima di introdurre o eliminare qualsiasi alimento dalla vostra alimentazione.
Fenilchetonuria ed aspartame
I pazienti con fenilchetonuria dovrebbero evitare i farmaci e gli alimenti che contengono l’aspartame, quindi ad esempio tutti quei cibi e bevande “light” spesso diffusi tra chi segue diete ipocaloriche. L’aspartame, quando è digerita, rilascia la fenilalanina rappresentando una fonte indiretta di quest’ultima.
Leggi anche:
- Le 5 migliori e dolci alternative allo zucchero bianco
- Il sorbitolo fa male alla salute? E’ causa di cattiva digestione?
- Xilitolo, eritritolo o stevia: quale dolcificante scegliere?
- Mannitolo: cos’è, diabete, lassativo, edema, struttura, fa male?
Latte in polvere per neonati con fenilchetonuria
Il latte in polvere artificiale normale ed il latte materno contengono la fenilalanina, quindi i neonati malati devono assumere un latte speciale che non contiene fenilalanina o comunque mescolarlo con quello artificiale o materno: in tal caso il medico calcola la quantità di latte materno o in polvere normale da mescolare con quello senza fenilalanina.
Svezzamento di un bimbo con fenilchetonuria
I genitori possono svezzare il bambino con alimenti a basso contenuto di fenilalanina negli stessi momenti previsti per lo svezzamento degli altri bambini. Questi alimenti vanno a sostituire la fenilalanina che il bambino assume tramite il latte materno o il latte in polvere e devono essere conteggiati dal medico nel consumo giornaliero di fenilalanina.
Sindrome fenilchetonurica e gravidanza
Le pazienti affette da PKU che riescono mediante l’adeguata dieta a raggiungere un’età fertile, se a dieta fin dall’inizio della gravidanza, non creano problemi rilevanti al feto che nascerà eterozigote per la PKU, ma sano. Seri problemi si hanno in gravidanze non programmate di donne PKU e, quindi, a dieta libera, dal momento che livelli elevati di fenilalanina ematici della madre possono avere effetti teratogeni sul feto, che ha quindi un elevato rischio di malformazioni anche gravi. Il feto rischia nei primi due mesi problemi cardiaci e successivamente microcefalia che può comportare un grave ritardo mentale. Durante il parto non si hanno problemi particolari. I rischi cardiaci e di microcefalia possono essere evitati unicamente programmando la gravidanza e mettendosi a dieta prima del concepimento. In questi casi non sembrano tornare utili farmaci come il Kuvan, la molecola sintetica del BH4 (sapropterina didroclorato). Questa molecola, utile in una forma molto rara della PKU in cui non è coinvolta l’attività dell’enzima p-fenilalanina idrossilasi ma, appunto la BH4, ha una qualche utilità nelle iperfenilalaninemie lievi o BH4 responsive, ma non è assolutamente in grado di controllare la fenilalanina ematica nelle forme di PKU classica. La dietoterapia è la unica soluzione per una gravidanza senza problemi per il feto. Durante la gravidanza, dal 4-5 mese in poi, la tolleranza alla fenilalanina aumenta progressivamente passando da valori, ad esempio, di 300 mg/die a 1400 mg/die al termine della gravidanza. Questo aumento della tolleranza è dovuto all’attività epatica del feto che, non essendo PKU (sarà un eterozigote per la malattia), è in grado di metabolizzare e smaltire sia la propria fenilalanina, sia quella in eccesso della madre.
Leggi anche:
- Le malattie genetiche più diffuse al mondo
- Differenza tra malattia genetica, ereditaria e congenita
- Malattia di Huntington: cos’è, ereditarietà, come si trasmette, età di insorgenza
- Sindrome di Klinefelter: cariotipo, cause, sintomi e cura
- Sindrome di Turner: cariotipo, cause, sintomi e segni caratteristici
- Sindrome di Down: cause, sintomi in gravidanza e nei neonati
- Sindrome di Marfan: trasmissione, sintomi e frequenza
- Malattia di Huntington: cos’è, ereditarietà, come si trasmette, età di insorgenza
- Differenza tra genetica ed epigenetica
- Differenza tra genetica e genomica
- Differenza genetica tra uomo e scimmia
- Cos’è un cromosoma ed a che serve?
- Differenza tra gene e allele
- Quanti cromosomi hanno esseri umani, scimmie, cani, gatti e topi?
- Quanti cromosomi ha chi è affetto da Sindrome di Down?
- Ectrodattilia: cause, cure ed immagini
- Polidattilia; cause, ereditarietà, sindromica e chirurgia
- Sindrome di Beals e padiglione auricolare “accartocciato”: sintomi e cure
- Differenza tra polidattilia, sindattilia, ectrodattilia, oligodattilia e aracnodattilia
- Dita ippocratiche congenite e secondarie: cause, sintomi e terapie
- Quante ossa ci sono nella mano e come si chiamano?
- Donne nate senza vagina: è la sindrome di Rokitansky-Kuster-Hauser
- Microtia ed anotia: la malformazione congenita dell’orecchio
- Palatoschisi: cause, problemi derivanti e cure
- Labbro leporino (cheiloschisi): tipi, cause, problematiche e cure
- Arinia: la mancanza congenita del naso
- Atrofia muscolare progressiva: cause, sintomi, cura, aspettativa di vita
- Differenze tra sclerosi laterale amiotrofica e sclerosi multipla
- Morbo di Parkinson: cause, sintomi, decorso, terapie
- Morbo di Alzheimer: cause, sintomi, decorso, terapie
- Demenza senile: cause, sintomi, decorso e cure
- Sindrome del cuore infranto: il falso infarto di chi ha il “cuore spezzato”
- Sindrome di Cornelia de Lange: avere l’aspetto di un bambino per tutta la vita
- Sindrome di Wolff-Parkinson-White: cos’è, cosa fare, come si cura
- Quando il paziente deceduto “resuscita”: la Sindrome di Lazzaro
- La sindrome che fa crescere il pene alle bambine di 12 anni
- Fibrosi cistica polmonare: cos’è, sintomi in neonati e bambini, cure
- La Sindrome da abbandono: cos’è e come si supera
- Anemia falciforme: cosa significa, cause, sintomi e cure
- Differenze tra la distrofia muscolare di Duchenne e di Becker
- Talassemia: cos’è, sintomi, cure, differenti tipi ed alimentazione
- Celiachia: cos’è il glutine, in quali alimenti è contenuto ed in quali no?
- Sindrome di Noonan: cause, sintomi nel neonato, aspettative di vita
- Sindrome di Bloom: cause, sintomi, diagnosi e terapia
- Insensibilità congenita al dolore: la strana malattia che non ti fa sentire nessun dolore
Lo staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o seguici su Twitter, su Instagram o su Pinterest, grazie!
