
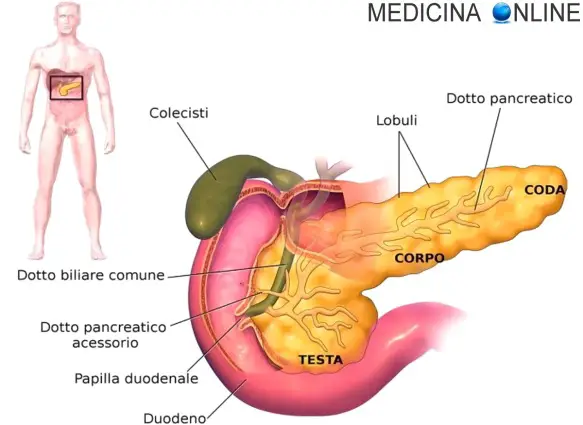
 Con “tumori del pancreas” si intende un gruppo di neoplasie benigne o maligne che si verificano in seguito alla moltiplicazione fuori controllo delle cellule pancreatiche. Il più diffuso tumore al pancreas è l’adenocarcinoma pancreatico, rappresentando l’85% dei casi, e il termine “cancro del pancreas” viene spesso usato come sinonimo proprio di questo tipo specifico di tumore pancreatico. L’adenocarcinoma pancreatico ha origine nelle cellule costituenti la porzione esocrina dell’organo deputata alla produzione di enzimi digestivi, zona dove si verificano quasi l’intera totalità dei tumori al pancreas. Meno diffuso (circa il 2% dei casi) è il tumore di tipo neuroendocrino, derivante dalla porzione endocrina del pancreas, cioè dalle cellule che producono gli ormoni pancreatici. Quest’ultimo tipo di tumore, oltre ad essere molto meno diffusi, sono anche molto meno aggressivi rispetto all’adenocarcinoma pancreatico.
Con “tumori del pancreas” si intende un gruppo di neoplasie benigne o maligne che si verificano in seguito alla moltiplicazione fuori controllo delle cellule pancreatiche. Il più diffuso tumore al pancreas è l’adenocarcinoma pancreatico, rappresentando l’85% dei casi, e il termine “cancro del pancreas” viene spesso usato come sinonimo proprio di questo tipo specifico di tumore pancreatico. L’adenocarcinoma pancreatico ha origine nelle cellule costituenti la porzione esocrina dell’organo deputata alla produzione di enzimi digestivi, zona dove si verificano quasi l’intera totalità dei tumori al pancreas. Meno diffuso (circa il 2% dei casi) è il tumore di tipo neuroendocrino, derivante dalla porzione endocrina del pancreas, cioè dalle cellule che producono gli ormoni pancreatici. Quest’ultimo tipo di tumore, oltre ad essere molto meno diffusi, sono anche molto meno aggressivi rispetto all’adenocarcinoma pancreatico.
Se sei qui per le recenti dichiarazioni del cantante Fedez, leggi anche: Fedez “Ho un raro tumore neuroendocrino del pancreas”. Ma le prospettive di sopravvivenza non sono così buone…
Tumori esocrini ed endocrini
I tumori del pancreas, come già prima anticipato, possono essere distinti in due grandi tipologie: esocrini ed endocrini.
Tumori esocrini del pancreas
I tumori esocrini originano dalla porzione esocrina del pancreas. Rappresentano il 98% di tutti i tumori del pancreas.
Il più diffuso è l’adenocarcinoma pancreatico (talvolta denominato anche “invasivo” e/o “duttale”), che rappresenta da solo l’85% di tutti i tumori del pancreas. Tale tumore origina in particolare nei condotti che trasportano le secrezioni esocrine pancreatiche (il succo pancreatico, ricco di enzimi digestivi) all’interno del duodeno.
Il secondo tipo più frequente è il carcinoma a cellule acinose del pancreas che origina dalle cellule che producono gli enzimi, comprendendo il 5% dei tumori del pancreas esocrino. Tali carcinomi possono causare un eccesso di produzione di alcune molecole, in questo caso enzimi digestivi, che talvolta comportano il verificarsi di sintomi paraneoplastici come eruzioni cutanee e dolori articolari.
Il cistoadenocarcinoma rappresenta solo l’1% dei tumori pancreatici e vanta una prognosi migliore rispetto agli altri tipi esocrini. Il pancreatoblastoma è una forma rara, tipica soprattutto dell’infanzia, con una prognosi relativamente buona. Altri tumori esocrini includono il carcinoma adenosquamoso, il carcinoma a cellule ad anello con castone, il carcinoma epatoide, il carcinoma colloide, il carcinoma indifferenziato e i carcinomi indifferenziati con cellule giganti tipo osteoclasti. Il tumore solido pseudopapillare è una rara neoplasia a basso grado che colpisce soprattutto le giovani donne ed ha generalmente una prognosi molto buona.
Leggi anche: Si può vivere senza pancreas? Conseguenze della pancreasectomia
Tumori endocrini (neuroendocrini) del pancreas
I tumori neuroendocrini originano dalla porzione endocrina del pancreas. Rappresentano il 2% di tutti i tumori del pancreas. La maggior parte dei tumori pancreatici di questo tipo sono i tumori neuroendocrini (NET), un gruppo eterogeneo di tumori benigni o maligni che derivano dalle cellule neuroendocrine del corpo, responsabili dell’integrazione tra il sistema nervoso e endocrino. I NET possono esordire nei vari organi del corpo, tra cui appunto il pancreas, e le varianti maligne sono considerate rare. I NET sono raggruppati in “funzionanti” e “non funzionanti”, a seconda della loro capacità di produrre gli ormoni. I tipi funzionanti secernono nel sangue ormoni come insulina, gastrina e glucagone, spesso in grandi quantità e dando luogo a sintomi gravi come l’ipoglicemia ma in questo modo tendezialmente si riesce ad ottenere una diagnosi relativamente precoce. I PanNET funzionanti più comuni sono gli insulinomi e i gastrinomi, dal nome degli ormoni che secernono. I tipi non funzionanti non secernono ormoni in quantità sufficiente a dare luogo a sintomi clinici evidenti. Per questo motivo, i PanNET non funzionanti sono spesso diagnosticati solo dopo che il tumore si è diffuso ad altre parti del corpo.
Leggi anche:
- Cos’è una ghiandola endocrina? A che servono gli ormoni ed il sistema endocrino?
- Differenza tra ghiandola esocrina ed endocrina con esempi
Diffusione e mortalità
Nel 2012 il tumore al pancreas ha provocato 330.000 decessi in tutto il mondo, un dato in costante crescita se raffrontato ai 310.000 decessi registrati nel 2010 e ai 200.000 del 1990. Anche se rappresenta solo il 2,5% dei nuovi casi di tumore diagnosticati, quello al pancreas è responsabile ogni anno per il 6% delle morti per cancro. È la settima causa di morte per cancro in tutto il mondo. Globalmente il cancro del pancreas è l’11° tumore più comune nelle donne e il 12° più comune negli uomini. La maggior parte dei casi registrati si verificano nei paesi sviluppati. La malattia è più comune negli uomini rispetto alle donne, anche se la differenza nei tassi si è ridotta negli ultimi decenni, riflettendo probabilmente l’aumento del tabagismo tra il genere femminile.
Segni e sintomi precoci
Nelle fasi iniziali della malattia, generalmente il paziente è asintomatico, cioè non ha alcun segno o sintomo della presenza della patologia. In alcuni casi la neoplasia può fornire sintomi aspecifici come:
- malessere;
- nausea;
- vomito;
- sintomi relativi a cattiva digestione.
Tali sintomi sono generalmente sottostimati dal paziente: in questa fase è quindi difficile che il tumore sia diagnosticato a meno che non sia riscontrato casualmente durante esami eseguiti per altri motivi, ad esempio una ecografia o una TAC addominale. In caso contrario la neoplasia progredisce e viene diagnosticata quando i sintomi, diventati più fastidiosi ed invalidanti, spingono il paziente a recarsi dal medico. Ciò si traduce in un frequente ritardo nella diagnosi, che spesso viene formulata solo quando il tumore è già in uno stadio avanzato e si è diffuso in altre parti del corpo. Questo è uno dei più importanti motivi alla base dell’elevata mortalità del tumore al pancreas: viene diagnosticato tardi, quando è spesso incurabile.
Segni e sintomi tardivi
I segni e i sintomi tardivi più comuni dell’affezione possono includere:
- un colorito giallo della pelle e delle sclere degli occhi (ittero);
- dolore addominale ;
- dolore alla schiena;
- sintomi di cattiva digestione;
- nausea;
- vomito;
- feci ipocoliche (di colore chiaro);
- sensazione di pienezza addominale;
- feci ricche di grasse e e maleodoranti;
- costipazione;
- diabete;
- urine scure;
- perdita di appetito;
- perdita di peso inspiegabile: è dovuta sia alla perdita di appetito in sé, sia alla perdita della funzione esocrina conseguente alla cattiva digestione;
- depressione;
- sindrome di Trousseau: caratterizzata da coaguli di sangue che si formano nei vasi sanguigni della vena porta, delle vene profonde degli arti o delle vene superficiali di qualsiasi parte del corpo (si riscontra in 1 paziente con tumore pancreatico su 10).
Ittero nel paziente con tumore pancreatico
L’ittero è generalmente dovuto ad una ostruzione dei dotti biliari causata dalla massa tumorale pancreatica (specie se interessa la testa del pancreas e non la coda) che tende a comprimerli. Per approfondire, leggi anche: Bilirubina diretta e indiretta: ittero, significato, patologie collegate Inoltre possono anche presentarsi (sempre a causa dei rapporti anatomici di quest’organo con quelli vicini) ostruzione duodenale, emorragia digestiva ed ascite; così come può sovrapporsi una pancreatite acuta al processo neoplastico.
Diabete nel paziente con tumore pancreatico
Circa la metà dei pazienti con adenocarcinoma pancreatico presenta il diabete al momento della diagnosi, condizione che può sfociare in iperglicemia fino alla glicosuria. Il diabete è considerato sia un fattore di rischio per il cancro al pancreas, ma anche causa di esso. Una recente insorgenza del diabete potrebbe essere considerata come un segno precoce della malattia. Gli individui di età superiore ai 50 anni che sviluppano il diabete, solitamente, hanno un rischio aumentato di otto volte di incorrere nell’adenocarcinoma del pancreas a tre anni dall’insorgenza, dopodiché il rischio relativo tende a calare.
Sintomi della diffusione del cancro pancreatico (metastasi)
La diffusione del tumore del pancreas ad altri organi (metastasi) può causare alcuni sintomi. In genere, l’adenocarcinoma pancreatico si diffonde prima ai linfonodi più vicini e poi al fegato o alla cavità peritoneale, all’intestino crasso o ai polmoni. Spesso, i pazienti con metastasi ad altri organi intra-addominali sviluppano ulteriori sintomi locali come ascite, masse addominali palpabili dall’esterno, epatomegalia e/o splenomegalia in caso di ostruzione della vena porta. Raramente si riscontra diffusione alle ossa o al cervello.
Leggi anche:
- Cancro al pancreas sempre più killer: casi aumentati del 60%
- Cos’è un tumore? Perché viene il cancro? Quali sono le cause?
- Pancreas: anatomia e funzioni in sintesi
- Insulina alta: cause, diabete, prediabete, valori normali e cure
Cause e fattori di rischio
Non esiste allo stato attuale della ricerca scientifica una causa specifica che determini un tumore pancreatico. Fattori di rischio noti sono:
- esposizione a radiazioni ionizzanti ad alto dosaggio (ad esempio quelle delle RX e TAC);
- famigliarità: tra il 5% e il 10% dei casi di tumore del pancreas ha una componente ereditaria;
- età > 40 anni (con ulteriore aumento del rischio > 70 anni: più della metà dei casi di adenocarcinoma pancreatico coinvolge ultrasettantenni;
- fumo di tabacco;
- obesità: un indice di massa corporea maggiore di 35 aumenta il rischio relativo di tumore pancreatico di circa il 50%;
- sesso maschile;
- pancreatite cronica;
- assunzione cronica di alcolici;
- esposizione cronica ad inquinanti ambientali, come acrilamide e insetticidi organico-clorurati;
- dieta ricca di : carni lavorate, carne rossa, carne cotta a temperature molto elevate (frittura, cottura alla griglia o barbecue), bevande zuccherate (soft drinks), bevande con fruttosio, eccesso di carboidrati e grassi;
- infezione da Helicobacter pylori;
- gengivite;
- malattia parodontale;
- diabete.
Malattie ereditarie che aumentano il rischio di tumore pancreatico
Il tumore al pancreas è correlato alle seguenti altre sindromi rare ereditarie:
- sindrome di Peutz-Jeghers, dovuta a mutazioni nel gene oncosoppressore STK11;
- sindrome del nevo displastico causa mutazioni nel gene CDKN2A soppressore del tumore;
- atassia-teleangectasia autosomica recessica;
- mutazioni autosomiche dominanti ereditate del gene BRCA2 e del gene PALB2;
- cancro colorettale ereditario non poliposico (sindrome di Lynch);
- poliposi adenomatosa familiare.
I PanNET sono spesso associati alla neoplasia endocrina multipla (MEN1) e alla sindrome di Von Hippel-Lindau.
Prevenzione
Non essendo note le cause dell’insorgenza di un tumore cerebrale, è impossibile dare consigli sulla prevenzione che valgano in modo assoluto. E’ certamente importante:
- evitare l’esposizione a radiazioni ionizzanti;
- evitare il fumo di sigaretta;
- alimentarsi in modo corretto;
- mantenere il peso corporeo entro i limiti di normalità;
- mantenere la glicemia entro limiti accettabili;
- non sottovalutare i sintomi prima elencati, anche se lievi, in individui a rischio (in tal caso è consigliabile lo screening).
In generale si consiglia di evitare i fattori di rischio modificabili precedentemente elencati. Per approfondire, leggi anche: Come prevenire i tumori ed il cancro? I 10 cambiamenti consigliati
Diagnosi
La diagnosi viene solitamente formulata grazie ad una combinazione di tecniche di imaging medicale, come l’ecografia o la tomografia computerizzata, esami del sangue e la biopsia (l’esame al microscopio di campioni di tessuto). Un eventuale screening della popolazione generale non si è dimostrato uno strumento efficace per la prevenzione.
Per approfondire, leggi anche:
- Marker CA 19-9 marcatore del tumore al pancreas
- Amilasi
- Lipasi
- Esami per valutare la funzionalità pancreatica
- Insufficienza pancreatica: esami, cause, sintomi, cura, dieta
- Ecografia addominale
- Colangiopancreatografia retrograda (ERCP)
- Biopsia pancreatica.
Cure in sintesi
La malattia può essere trattata mediante la chirurgia, la radioterapia, la chemioterapia, le cure palliative o con una combinazione di queste. La scelta del trattamento è in parte basata sulla sede della neoplasia e sulla stadiazione clinica. La chirurgia è l’unico trattamento curativo, ma può essere intrapresa anche nel tentativo di migliorare la qualità della vita, senza perseguire l’obiettivo della cura. La duodenocefalopancresectomia è una delle opzioni chirurgiche più complesse. Il ricorso alle cure palliative è consigliato anche per coloro che ricevono un trattamento finalizzato alla guarigione.
Leggi anche:
- Duodenocefalopancresectomia: complicanze, dieta, sopravvivenza
- Chemioterapia: durata, in pastiglie, come funziona, fa male, perché farla?
- E’ più “pesante” la chemioterapia o la radioterapia?
- Come funziona, si somministra e si svolge la radioterapia?
Prognosi e sopravvivenza in sintesi
Ogni anno l’insieme dei tumori pancreatici determina oltre 350 mila morti in tutto il mondo, rappresentando la settima causa più comune di morte per tumore, con una maggiore prevalenza nel mondo sviluppato. In genere l’adenocarcinoma pancreatico, che come abbiamo visto è il tipo più diffuso, è caratterizzato da una prognosi spesso infausta: dopo la diagnosi, ad un anno vi è una sopravvivenza del 25% ed al di sotto del 5% a cinque anni (circa l’1% in caso di stadio 4). L’aspettativa di vita per un paziente con adenocarcinoma inoperabile è inferiore a 5 anni per il 99% dei pazienti. Per casi diagnosticati precocemente, il tasso di sopravvivenza a cinque anni sale al 14% circa. I tumori neuroendocrini hanno risultati migliori; a cinque anni dalla diagnosi, il 65% dei pazienti è in vita, anche se tali dati variano notevolmente a seconda del tipo di tumore.
Per approfondire leggi il paragrafo “Prognosi” più in fondo nell’articolo.
Stadiazione TNM del tumore pancreatico
Il tumore al pancreas viene solitamente valutato soprattutto tramite tomografia computerizzata. Il sistema di stadiazione più diffuso per questo tumore è quello proposto dalla Joint Committee on Cancer (AJCC), insieme con l’Union for International Cancer Control (UICC). Il sistema AJCC-UICC indica quattro fasi generali principali, che vanno dallo stadio primitivo fino a quello relativo alla malattia avanzata. Tale sistema si basa sulla classificazione TNM in cui si considerano le dimensioni del tumore (T), il possibile coinvolgimento dei linfonodi (N) e l’eventuale presenza di metastasi (M). Per facilitare la decisione circa il possibile trattamento, i tumori sono divisi in tre categorie più ampie, a seconda che la rimozione chirurgica possa apparire possibile. In questo modo, i tumori sono giudicati “resecabili”, “resecabili borderline” o “non resecabili”. Quando la malattia è ancora in una fase iniziale (AJCC-UICC fasi I e II), senza che vi sia un coinvolgimento dei grandi vasi sanguigni o di organi distanti come il fegato o i polmoni, la resezione chirurgica del tumore può normalmente essere eseguita, a condizione che il paziente sia disposto a sottoporsi ad un intervento così complesso e si ritenga che sia sufficientemente in salute per affrontarlo. Il sistema di stadiazione AJCC-UICC consente la distinzione tra lo stadio III dei tumori che vengono giudicati “resecabili borderline” (dove la chirurgia è tecnicamente possibile perché il tronco celiaco e l’arteria mesenterica superiore sono ancora liberi dalla malattia) e quelli che sono “non resecabili” (cioè non operabili, a causa di una malattia localmente avanzata); in termini di classificazione più dettagliata TNM, questi due gruppi corrispondono rispettivamente allo stadio T3 e T4. In parole povere i tumori pancreatici:
- T1 e T2 sono operabili;
- T3 potrebbero essere operabili o non;
- T4 non sono operabili.
Di seguito è riportata la classificazione TNM:
Stadio Descrizione
T0 non evidenza di neoplasia
Tis tumore in situ
T1 limitato al pancreas e con diametro minore di 2 cm
T2 come 1 ma con diametro maggiore di 2 cm
T3 oltre il pancreas senza coinvolgere tripode
celiaco/arteria mesenterica superiore
T4 coinvolge le strutture citate sopra
N0 assenza di metastasi linfonodali
N1 presenza di metastasi linfonodali
M0 assenza di metastasi a distanza
M1 presenza di metastasi a distanza
-
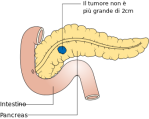
- Stadio T1
-
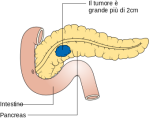
- Stadio T2
-
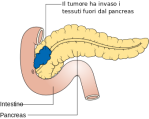
- Stadio T3
-
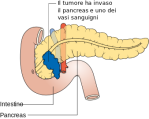
- Stadio T4 (inoperabile)
-
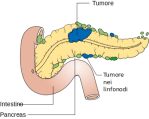
- Coinvolgimento linfonodale L1
-
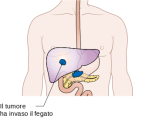
- Presenza di metastasi M1
Gli stadi risultanti possibili sono:
Stadio 0: Tis,N0,M0
Stadio 1A: T1,N0,M0
Stadio 1B: T2,N0,M0
Stadio 2A: T3,N0,M0
Stadio 2B: T1-3,N1,M0
Stadio 3: T4,N(qualunque),M0
Stadio 4: T e N(qualunque),M1
Se ne deduce che:
- La presenza di coinvolgimento linfonodale (L1) colloca qualsiasi tumore pancreatico almeno nello stadio 2B (a patto che NON ci siano metastasi).
- La presenza di un tumore pancreatico di grosse dimensioni (T4) colloca qualsiasi tumore almeno in stadio 3, a prescindere dal coinvolgimento linfonodale ed a patto che NON ci siano metastasi.
- La presenza di metastasi (M1), colloca qualsiasi tumore pancreatico in stadio 4, a prescindere dalle dimensioni della massa tumorale e dal coinvolgimento linfonodale.
Leggi anche:
- Stadiazione e classificazione TNM: cancro curabile o terminale?
- Cosa sono le metastasi? Tutti i tumori danno metastasi?
Trattamento
Trattamento dei tumori esocrini
A seguito della diagnosi, si procede con la valutazione circa la possibilità di eseguire un intervento chirurgico di resezione, l’unico trattamento che possa essere curativo. La decisione è vincolata soprattutto dal grado di diffusione del tumore, ma anche la posizione esatta della massa neoplastica è un fattore significativo e le immagini ottenute tramite tomografia computerizzata (TC) possono mostrare il suo rapporto anatoomico con i principali vasi sanguigni. Anche la condizione di salute generale del paziente deve essere valutata, tuttavia l’età di per sé non rappresenta un ostacolo alla chirurgia. Se la prospettiva chirugica non è possibile, la chemioterapia e, in misura minore, la radioterapia sono approcci che possono essere proposti nella maggioranza dei casi. Gli specialisti consigliano di affidare la gestione di un tumore pancreatico ad un gruppo multidisciplinare di cui facciano parte specialisti in diversi ambiti dell’oncologia e quindi sono da preferire i centri di riferimento più grandi.
Trattamento dei PanNET
Il trattamento dei PanNET, inclusi i tipi maligni meno comuni, può includere una serie di approcci. Alcuni piccoli tumori inferiori a 1 cm, che spesso vengono identificati casualmente, ad esempio durante una tomografia computerizzata eseguita per altri scopi, possono essere gestiti attraverso una vigile attesa. Questo dipende dalla valutazione del rischio chirurgico, correlato dalla posizione del tumore e alla presenza di eventuali altri problemi medici. I tumori unici all’interno del pancreas (tumori localizzati) o con limitate metastasi, ad esempio al fegato, possono essere rimossi a seguito di un intervento chirurgico. Il tipo di intervento dipende dalla localizzazione del tumore e dal livello di diffusione linfonoidale. Per i tumori localizzati, le procedure chirurgiche possono essere nettamente meno invasive rispetto a quelle utilizzate per il trattamento dell’adenocarcinoma pancreatico, altrimenti sono molto simili a quelle per i tumori esocrini. I possibili risultati possono variare notevolmente: alcune tipologie hanno un tasso di sopravvivenza molto alto dopo l’intervento chirurgico, mentre altre hanno un decorso meno positivo. Essendo che queste situazioni si presentano abbastanza raramente, le linee guida sottolineano che il trattamento debba essere effettuato in un centro specializzato. Nei casi di presenza di metastasi epatiche può essere considerato il ricorso al trapianto di fegato. Per i tumori endocrini (o “funzionanti”), i farmaci analoghi alla somatostatina, come l’octreotide, sono in grado di ridurre l’eccessiva produzione di ormoni. Il lanreotide può rallentare la crescita del tumore. Se il tumore non è suscettibile di rimozione chirurgica ed è causa di sintomi, la terapia mirata con everolimus o sunitinib può ridurre le sue manifestazioni e rallentare la progressione della malattia. La chemioterapia citotossica standard non è in genere molto efficace per il trattamento dei PanNET, ma può essere utilizzata quando altri trattamenti farmacologici non riescono a prevenire la progressione della malattia o nel caso di tumori PanNET scarsamente differenziati. La radioterapia viene talvolta utilizzata se vi è dolore dovuto alla compressione degli oegani adiacenti consegente all’estensione della massa tumorale, come ad esempio nelle metastasi alle ossa. Alcuni PanNET assorbono peptidi o ormoni specifici e possono rispondere alla terapia di medicina nuclearecon peptidi radiomarcati o ormoni come lo iobenguane (iodio-131-MIBG). L’ablazione a radiofrequenza (RFA), può essere utilizzata anche con la crioablazione e l’embolizzazione dell’arteria epatica.
Cure palliative nel malato inoperabile e terminale
Le cure palliative sono cure mediche che si concentrano sul trattamento dei sintomi di una malattia grave, come può essere il tumore, al fine di migliorare la qualità della vita. Siccome l’adenocarcinoma pancreatico viene solitamente diagnosticato quando si trova già ad uno stadio avanzato, le cure palliative sono spesso l’unico trattamento possibile. Esse non si focalizzano sul trattamento del tumore stesso, ma sulla gestione dei sintomi, come il dolore e la nausea, aiutando nel processo decisionale di approccio alla malattia. Il dolore può essere gestito tramite farmaci, come gli oppioidi o attraverso alcune procedure interventistiche, come un blocco nervoso sul plesso celiaco. Ciò altera, o distrugge (a seconda della tecnica utilizzata), i nervi che trasmettono il dolore. Tale intervento è un modo sicuro ed efficace per ridurre il dolore, riducendo generalmente la necessità di utilizzare analgesici oppioidi che presentano significativi effetti collaterali negativi. Altri sintomi o complicazioni che possono essere trattati con la chirurgia palliativa sono le ostruzioni dovute alla massa tumorale dell’intestino o dei dotti biliari. Per questi ultimi, che vengono compromessi in più della metà dei casi, un piccolo tubo metallico, chiamato stent può essere inserito tramite procedura endoscopica per mantenere i condotti pervi. Le cure palliative possono anche aiutare nella gestione della depressione clinica che spesso si accompagna alla diagnosi di una neoplasia pancreatica. Sia la chirurgia che i tumori inoperabili in fase avanzata, spesso portano a disturbi del sistema digerente per colpa di una mancanza di prodotti esocrini del pancreas (insufficienza esocrina). Questi possono essere trattati assumendo pancreatina che contiene enzimi pancreatici. La difficoltà nello svuotamento dello stomaco è frequente e può rappresentare un problema serio, che comporta l’ospedalizzazione. Il trattamento può comportare una varietà di approcci, tra cui lo svuotamento dello stomaco mediante aspirazione nasogastrica e l’assunzione di farmaci chiamati inibitori della pompa protonica o antagonisti H2, i quali sono in grado di ridurre la produzione di succo gastrico.
Prognosi
| Stadiazione clinica | Sopravvivenza a cinque anni | |
|---|---|---|
| Tumori pancreatici esocrini | Tumori neuroendocrini trattati con la chirurgia | |
| IA / I | 14% | 61% |
| IB | 12% | |
| IIA / II | 7% | 52% |
| IIB | 5% | |
| III | 3% | 41% |
| IV | 1% | 16% |
Tipicamente, i pazienti con adenocarcinoma pancreatico o altri tumori esocrini meno comuni, non hanno una buona prognosi, in parte perché spesso la diagnosi viene formulata quando il tumore è già localmente avanzato o si è diffuso ad altre parti del corpo. Risultati migliori si hanno nel caso dei PanNET, molti dei quali sono benigni e non presentano sintomi clinici e i casi non trattabili con la chirurgia hanno un tasso medio di sopravvivenza a cinque anni del 16%, anche se le prospettive variano notevolmente a seconda del tipo. Per l’adenocarcinoma pancreatico localmente avanzato e quello metastatico, che insieme rappresentano oltre l’80% dei casi, numerosi studi recenti mirati a confrontare i diversi regimi chemioterapici, hanno mostrato un aumento della sopravvivenza, ma non superiore all’anno. Negli Stati Uniti, la sopravvivenza globale a cinque anni per il tumore pancreatico è migliorata negli anni, passando dal 2% nei casi diagnosticati tra il 1975 e il 1977, al 4% tra quelli rilevati dal 1987 al 1989, al 6% tra quelli scoperti dal 2003 al 2009. In meno del 20% dei casi di adenocarcinoma del pancreas con diagnosi di una crescita cancerosa localizzata e di scarse dimensioni (meno di 2 cm in fase T1), circa il 20% degli statunitensi sono sopravvissuti a cinque anni.
Leggi anche:
- Capire se si ha un tumore: come viene diagnosticato un cancro
- Come nasce un cancro? Cosa sono i cancerogeni e come avviene la cancerogenesi?
- Glucagone: cos’è, a cosa serve, alto, adrenalina e diabete
- Somatostatina: cos’è ed a cosa serve? Efficacia come farmaco antitumorale
- Differenza tra insulina e glucagone nella regolazione della glicemia
- Differenza tra cancro e carcinoma con esempi
- Differenze tra il diabete di tipo 1 e 2 (insulino dipendente e resistente)
- Emoglobina glicata alta, valori normali, IFCC e diabete
- Diabete: quale frutta mangiare e quale evitare? Guida completa
- Alimentazione consigliata a chi soffre di diabete: i cibi che tengono sotto controllo la glicemia
- Differenza tra prevenzione primaria, secondaria e terziaria con esempi
- Differenza tra chemioterapia adiuvante e neoadiuvante
- Differenza tra chemioterapia sistemica e loco-regionale
- Chemioterapia: gli effetti collaterali più e meno comuni
- Chemioterapia: durata effetti collaterali ed effetti di lunga durata
- La chemioterapia può provocare un secondo tumore?
- La chemioterapia uccide? Il lato oscuro della terapia antitumorale
- Chemioterapia senza caduta capelli: ecco la cuffia per non perderli
- Quando non fare chemioterapia? Le controindicazioni
- Chemioterapia in gravidanza: può far male al bambino?
- Differenza tra remissione parziale, totale e guarigione
- Differenza tra tumore benigno, maligno, neoplasia, cancro e metastasi
- Differenza tra adenocarcinoma e carcinoma con esempi
- Gli uomini possono avere il tumore al seno?
- Differenza tra metaplasia, displasia e neoplasia con esempi
- Differenza tra tumore e tessuto normale con esempi
- Cancro al seno: sintomi precoci, diagnosi, terapia e prevenzione
- Differenza tra ipertrofia ed iperplasia con esempi
- Differenza tra iperplasia e neoplasia
- Differenza tra atrofia, distrofia ed aplasia con esempi
- Esofago di Barrett: sintomi iniziali, diagnosi, terapia, dieta e chirurgia
- Tumore del colon retto: diagnosi, metastasi, prognosi e stadiazione
- Tumore del colon retto: sintomi iniziali, tardivi e ritardo nella diagnosi
- Tumore del colon retto: trattamento chirurgico, radioterapia e chemioterapia
- Tumore del colon retto con metastasi: chirurgia, chemioterapia e terapie biologiche
Dott. Emilio Alessio Loiacono
Medico Chirurgo
Direttore dello Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o unisciti al nostro gruppo Facebook o ancora seguici su Twitter, su Instagram, su Mastodon, su YouTube, su LinkedIn, su Tumblr e su Pinterest, grazie!
