
 Il morbo di Addison (anche chiamata malattia di Addison, insufficienza surrenalica cronica, ipocortisolismo, iposurrenalismo o Addison’s disease) è una malattia endocrina cronica corrispondente alla forma primitiva di insufficienza corticosurrenale cronica, correlata ad una severa, permanente ed irreversibile riduzione della secrezione degli ormoni corticosteroidi secreti dalla corteccia del surrene. Ricordiamo che la corteccia surrenale è la parte più esterna del surrene e produce gli ormoni steroidei (corticosteroidi ed ormoni sessuali). E’ divisa in tre zone:
Il morbo di Addison (anche chiamata malattia di Addison, insufficienza surrenalica cronica, ipocortisolismo, iposurrenalismo o Addison’s disease) è una malattia endocrina cronica corrispondente alla forma primitiva di insufficienza corticosurrenale cronica, correlata ad una severa, permanente ed irreversibile riduzione della secrezione degli ormoni corticosteroidi secreti dalla corteccia del surrene. Ricordiamo che la corteccia surrenale è la parte più esterna del surrene e produce gli ormoni steroidei (corticosteroidi ed ormoni sessuali). E’ divisa in tre zone:
- zona glomerulare: è la parte più esterna della corticale del surrene e produce mineralcorticoidi come l’aldosterone che controlla i livelli di elettroliti e la quantità di acqua presenti nel sangue;
- zona fascicolata: è la parte centrale della corticale del surrene e produce glucocorticoidi come il cortisolo che controlla il metabolismo di carboidrati, lipidi e proteine;
- zona reticolata: è la parte più interna della corticale del surrene e produce soprattutto androgeni (ormoni sessuali maschili come il testosterone) e piccole quantità di estrogeni (ormoni sessuali femminili).
Gli ormoni prodotti nella parte corticale del surrene sono coinvolti in una varietà di meccanismi fisiologici, inclusi quelli che regolano l’infiammazione, il sistema immunitario, il metabolismo dei carboidrati, delle proteine, il livello di elettroliti nel sangue, la sessualità e la riproduzione.
Il morbo di Addison è caratterizzato da una serie di sintomi relativamente non specifici, come dolore addominale e debolezza, ma in determinate circostanze, questi possono evolvere con attacchi acuti gravi che possono comportare una severa ipotensione e il coma. La condizione deriva da problemi che interessano direttamente la ghiandola surrenale che possono essere dovuti a un malfunzionamento del sistema immunitario, da alcune infezioni o da varie cause più rare. In caso di situazioni particolarmente stressanti un morbo di Addison teoricamente controllato, può evolvere in una crisi addisoniana potenzialmente fatale. La condizione prende il nome dal dott. Thomas Addison che per primo ne descrisse la condizione nel 1849.
Insufficienza corticosurrenale primaria cronica, acuta, secondaria e terziaria
La malattia di Addison è conosciuta anche come insufficienza corticosurrenale primaria cronica, per distinguerla dall’insufficienza corticosurrenale primaria acuta, causata dalla sindrome di Waterhouse-Friderichsen, in cui l’insufficienza surrenalica è determinata da infezione batterica da Neisseria meningitidis o altro patogeno. La malattia di Addison è anche distinta dall’insufficienza surrenalica secondaria e terziaria:
- insufficienza surrenalica secondaria: la scarsa funzione del surrene è dovuta a scarsa secrezione di ACTH da parte dell’ipofisi;
- insufficienza surrenalica terziaria: la scarsa funzione del surrene è dovuta a scarsa secrezione di CHR da parte dell’ipotalamo.
Per approfondire, leggi anche:
- Ormone adrenocorticotropo o corticotropina (ACTH): cos’è e quali sono le sue funzioni
- Ormone di rilascio della corticotropina (CRH): produzione e funzioni
- Asse ipotalamo-ipofisi-surrene: funzionamento ed ormoni rilasciati
Diffusione
La prevalenza è di 1 su 8.000 ed è in aumento grazie al miglioramento della diagnosi, colpisce prevalentemente gli adulti tra 30 e 50 anni e soprattutto donne. Le cause della malattia vanno identificate in tutte quelle condizioni morbose che comportino la distruzione quasi completa (oltre 90%) della corteccia surrenalica.
Cause
Circa il 70% dei casi di malattia di Addison è dovuto ad aggressione auto-immunitaria della ghiandola (atrofia surrenalica autoimmune o atrofia idiopatica della corteccia surrenale). Questa forma di malattia di Addison è caratterizzata da un infiltrato linfomonocitario (adrenalite) della corteccia surrenale. In circolo sono presenti ACA (Anti-Cortex Antibodies) diretti verso la 21 idrossilasi, enzima chiave della steroidogenesi. Questi anticorpi sono molto specifici e occasionalmente si possono trovare anche in pazienti affetti da malattia di Basedow, tiroidite di Hashimoto e Iddm e la loro presenza indica il rischio di sviluppo di insufficienza surrenalica. La rilevazione di questi anticorpi può precedere di anni l’insorgenza di un’insufficienza surrenalica manifesta. Il primo bersaglio sembra essere la zona glomerulare produttrice di aldosterone; infatti si osserva all’inizio un’elevazione dell’attività reninica del siero. Gli anticorpi in seguito alla distruzione del surrene e quindi all’instaurarsi della sintomatologia clinica scompaiono. La forma autoimmunitaria di Addison si può presentare in 3 forme:
- associata a sindrome pluriendocrina di tipo I: (ipoparatiroidismo, insufficienza surrenale, ipogonadismo primitivo, candidosi mucocutanea, anemia perniciosa);
- associata a sindrome pluriendocrina di tipo II: chiamata anche sindrome di Schmidt (insufficienza surrenale, tireopatie autoimmuni, diabete mellito insulino-dipendente);
- forma isolata.
Risulta quindi importante la ricerca di altri autoanticorpi rivolti verso altri tipi di strutture ghiandolari. In 20-25% dei casi di malattia di Addison è la conseguenza della distruzione della ghiandola da parte di granulomi soprattutto da TBC (che è divenuto recentemente più frequente nei paesi in via sviluppo), in casi rari Addison è dovuto a lesioni distruttive della ghiandola da parte di tumori primitivi o metastatici del surrene (linfomi), emorragie o infarti surrenali, amiloidosi, infezioni fungine e infezioni da CMV (soprattutto in AIDS). Fra le cause minori ricordiamo anche ipoplasia congenita dei surreni, malattia a carattere ereditario caratterizzata dalla mancata responsività delle cellule surrenali all’ACTH e malattia di Addison di origine iatrogena, da surrenectomia bilaterale, in pazienti con forme particolarmente gravi di malattia di Cushing.
Fisiopatologia
La carenza di aldosterone e cortisolo è responsabile delle manifestazioni più gravi della malattia di Addison, mentre la carenza di androgeni surrenalici è solo responsabile della riduzione dell’apparato pilifero nella donna, specie a livello ascellare.
Carenza di aldosterone
La carenza di aldosterone determina una ridotta capacità di trattenere sodio (quindi anche di acqua) e di eliminare potassio a livello dei tubuli renali, perciò nella malattia di Addison si assiste all’aumento dell’escrezione di sodio e alla diminuzione dell’escrezione di potassio nelle urine che sono diluite: ne risultano basse concentrazioni ematiche di sodio e di cloro e un’alta concentrazione sierica di potassio. L’incapacità di concentrare le urine, associata alle modificazioni dell’equilibrio elettrolitico, determina una grave disidratazione, ipertonicità plasmatica, acidosi, ipovolemia, perdita di peso, ipotensione, diminuzione della gettata cardiaca, astenia intensa e facile insorgenza di episodi lipotimici da ipotensione ortostatica, inoltre iperpotassiemia indotta da carenza di aldosterone può essere responsabile di disturbi del ritmo (asistolia, blocchi A-V, ecc.) L’acido glicirretico possiede una molecola di struttura simile a quella dell’aldosterone (affinità bassa, ma definita, dunque che richiede alti dosaggi per avere un’attività biologica), un meccanismo d’azione e sintomatologia simili (tanto che l’ipopotassiemia da liquirizia era una delle cause più frequenti di ipertensione nei Paesi dove la sua disponibilità non era limitata), un “agonismo” diretto acido glicirretico-aldosterone che permetteva a pazienti affetti dalla malattia di Addison di sopravvivere solo assumendo dosi elevate di radici di liquirizia: la carenza di aldosterone faceva aumentare il potassio e perdere acqua e sodio, mentre la liquirizia operava in modo contrario.
Carenza di cortisolo
Il deficit di cortisolo contribuisce all’ipotensione e provoca disturbi metabolici come ridotta gliconeogenesi, diminuzione di glicogeno epatico, diminuita mobilizzazione e utilizzazione dei grassi, ipoglicemia che insieme all’iponatriemia sono responsabili dell’intensa astenia e della perdita di peso che caratterizza i pazienti addisoniani. Le alterazioni metaboliche vengono ritenute responsabili anche dei disturbi psichici che sono rilevabili clinicamente in circa 70% di questi pazienti e che consistono principalmente in apatia, ridotto interesse verso l’ambiente, depressione. Il cortisolo esercita normalmente un’azione stimolante l’eritropoiesi e influenza il traffico dei leucociti tra il compartimento intravascolare e i tessuti, inducendo aumento dei granulociti e diminuzione dei linfociti e degli eosinofili, la sua carenza determina modificazioni in senso opposto degli elementi ematici con anemia, neutropenia, linfocitosi ed eosinofilia. La riduzione dei livelli ematici di cortisolo comporta ipersecrezione di ACTH ipofisario e dei peptidi correlati come beta-lipotropina, alfa e beta-MSH, i quali hanno attività melanocito-stimolante e producono iperpigmentazione della cute e della mucosa caratteristica della malattia di Addison. La conseguenza più grave della mancanza di cortisolo è comunque rappresentata dall’incapacità dei pazienti addisoniani di rispondere adeguatamente a ogni tipo di stress fisiologico e patologico, questi soggetti risultano estremamente fragili di fronte a eventi morbosi, traumi, interventi chirurgici che sono agevolmente superati da soggetti normali.
Sintomi
Nella forma conclamata la malattia si esprime sintomatologicamente con una triade caratteristica: astenia, melanodermia, ipotensione. La progressione della malattia è lenta e graduale in rapporto alla progressione delle lesioni distruttive della ghiandola. Quando la perdita di tessuto surrenalico supera il 90% si ha un quadro completo di insufficienza surrenalica cronica. I principali sintomi e segni sono:
- astenia, ipoglicemia, affaticabilità e ipotensione ortostatica sono sintomi precoci, all’inizio l’astenia si manifesta dopo un periodo di stress, mentre successivamente il paziente è costretto a rimanere a letto. Al pari dell’astenia, anoressia e la perdita di peso sono elementi clinici costantemente presenti nell’Addison tanto che tale diagnosi in un paziente che goda di buon appetito o che presenti un incremento ponderale o che denunci un buon vigore fisico è improbabile;
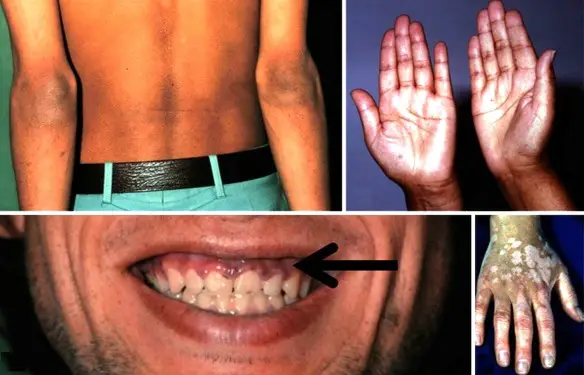
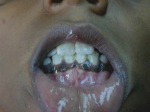
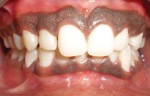
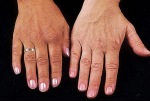
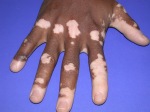
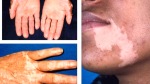
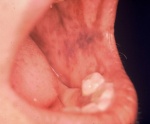
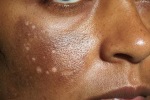
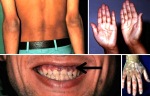
- l’acantosi nigricans e la melanodermia, vale a dire iperpigmentazione cutanea, costituiscono la seconda manifestazione cardinale della malattia e sono dovute a ipersecrezione di ACTH, ormone che deriva dallo stesso precursore dell’MSH (ormone che stimola i melanociti). Tale precursore è detto proopiomelanocortina (POMC). La iperpigmentazione nelle fasi iniziali è lieve e può essere confusa con residui di un’abbronzatura o con un naturale colorito bruno della pelle. Alcune caratteristiche dell’iperpigmentazione addisoniana permettono di distinguerla da altre forme: essa è più evidente a livello delle pieghe cutanee (specie alle pliche dei palmi delle mani), nelle zone esposte a pressione o a continui attriti (come nel solco sottomammario nelle donne che portano il reggiseno, o in corrispondenza del colletto, della cintura dei pantaloni, dei gomiti e delle ginocchia) e in tutte le regioni normalmente iperpigmentate (come areole mammarie, la regione scrotale e perineale) e ancora in sede di cicatrici prodotte successivamente all’esordio della malattia. Inoltre nella malattia di Addison la iperpigmentazione si estende anche alle mucose, fra queste quelle più frequentemente colpite sono la mucosa orale in corrispondenza delle guance, delle gengive, del palato, la mucosa vaginale e quella rettale. L’associazione fra melanodermia cutanea e iperpigmentazione mucosa è fortemente suggestiva per la natura addisoniana delle deviazioni pigmentarie. Va tuttavia aggiunto che fra le zone di iperpigmentazione cutanee possono essere intercalate delle chiazze di ipopigmentazione che possono giungere sino alla vitiligine. La cute è particolarmente secca per la disidratazione dovuta alla deplezione sodica;
- vertigini;
- attacchi sincopali;
- turbe neuropsichiche come irritabilità, ansia e apatia, nonché difficoltà di concentrazione;
- dolori addominali, soprattutto in regione epigastrica con nausea, vomito e diarrea (diagnosi differenziale con ulcera peptica);
- alterazioni della funzione sessuale sono piuttosto frequenti, l’amenorrea, la più comune di esse, può essere la conseguenza della perdita di peso cui va incontro il paziente addisoniano o esprimere la concomitanza di una insufficienza gonadica primitiva a genesi autoimmune. La perdita dei peli pubici e ascellari nella donna esprime la ridotta secrezione degli androgeni surrenalici.
Sintomi di crisi surrenalica
La crisi surrenalica è caratterizzata da:
- astenia profonda;
- forte dolore a livello addominale, in regione lombare o alle gambe;
- collasso vascolare periferico;
- blocco renale con iperazotemia.
La temperatura corporea può essere bassa, sebbene possa spesso manifestarsi una febbre grave, in particolare quando la crisi è scatenata da un’infezione acuta. Un significativo numero di pazienti con una perdita parziale della funzione surrenalica (riserva corticosurrenalica limitata) appare in buona salute, ma subisce crisi surrenaliche se sottoposti a stress fisiologici (come interventi chirurgici, infezioni, ustioni gravi e patologie gravi).
Diagnosi e diagnosi differenziale
La diagnosi viene sospettata sulla base dei sintomi e dei segni e confermata dai test di laboratorio. L’insufficienza corticosurrenalica può essere diagnosticata dimostrando l’incapacità ad aumentare i livelli plasmatici di cortisolo o l’escrezione urinaria di cortisolo libero dopo la somministrazione di ACTH.
Test di valutazione per l’insufficienza corticosurrenalica: il test si esegue iniettando 250 microgrammi di ACTH sintetico (Cortrosyn) EV. Prima dell’iniezione il cortisolo plasmatico normale è compreso fra 5 e 25 microgrammi/dL (fra 138 e 690 nmol/L) e raddoppia fra i 30 e i 90 minuti, con un minimo di 20 microgrammi/dL (552 nmol/L). I pazienti con malattia di Addison hanno valori bassi o normali che non subiscono incrementi.
Un test di stimolo all’ormone adrenocorticotropo (ACTH) prolungato (prelievi eseguiti nel corso di 24 h), può essere impiegato per diagnosticare un’insufficienza surrenalica secondaria (o terziaria, ossia, ipotalamica). Viene somministrato 1 mg IM di cosintropina, e vengono misurati i livelli di cortisolo a diversi intervalli nel corso di 24 h, tipicamente a 1,6, 12 e 24 h. I risultati della prima ora sono simili sia per il test breve (in cui i prelievi vengono sospesi dopo 1 h) che per quello prolungato, ma nel morbo di Addison non si manifesta un ulteriore incremento dopo i 60 min. Nell’insufficienza surrenalica secondaria e terziaria, invece, i livelli di cortisolo continuano ad aumentare dopo le ≥ 24 h. Unicamente nei casi di atrofia surrenalica prolungata, è necessaria una premedicazione surrenalica (con ormone adrenocorticotropo ACTH a lunga durata d’azione). Di solito si comincia con il test breve, dal momento che una risposta normale esclude la necessità di ulteriori indagini.
Distinzione tra insufficienza corticosurrenalica primitiva e secondaria: la maggior parte dei casi di ipocorticosurrenalismo secondario è provocata dalla distruzione dell’ipofisi. La TC o la RMN della sella possono quindi essere utili per escludere la presenza di tumori o atrofia. Nei pazienti con una patologia primitiva del surrene, il livello plasmatico di ACTH è elevato (> 50 pg/mL). I pazienti con insufficienza ipofisaria o con deficit isolato di ACTH hanno un basso livello dell’ormone. Se si sospetta un’insufficienza surrenalica secondaria, questa potrà essere confermata da test al metirapone. Il metirapone è un farmaco in grado di bloccare l’enzima 11-b-idrossilasi, quindi, deprime la conversione dell’11-desossicortisolo in cortisolo, i cui livelli plasmatici si riducono con conseguente incremento della secrezione di ACTH.
L’iperpigmentazione può derivare da un carcinoma broncogeno, dall’ingestione di metalli pesanti (p.es., ferro o argento), da patologie croniche della cute o dall’emocromatosi. Anche la sindrome di Peutz-Jeghers è caratterizzata da una pigmentazione della mucosa buccale e rettale. Spesso l’iperpigmentazione è associata alla vitiligine e questo può suggerire il morbo di Addison, sebbene anche altre malattie possano provocare questa associazione.
La debolezza che deriva dal morbo di Addison scompare con il riposo, a differenza delle forme neuropsichiatriche che, spesso, sono peggiori al mattino piuttosto che dopo un periodo di attività. La maggior parte delle miopatie, che determinano debolezza, può essere esclusa dalla diagnosi differenziale in base alla loro distribuzione, all’assenza di pigmentazione anomala e ai caratteristici rilievi di laboratorio.
I pazienti con insufficienza surrenalica sviluppano ipoglicemia a digiuno, a causa della diminuita gluconeogenesi. Al contrario, i pazienti con ipoglicemia da ipersecrezione di insulina, possono avere crisi in ogni momento; di solito mostrano un appetito aumentato associato ad aumento di peso e presentano una normale funzione surrenalica.
I bassi livelli sierici di Na causati dal morbo di Addison devono essere distinti da quelli presenti nei pazienti edematosi con patologie cardiache o epatiche (specialmente quelli che assumono diuretici), dall’iponatriemia da diluizione, caratteristica della sindrome da inappropriata secrezione di ormone antidiuretico (ADH) e dalla nefrite sale disperdente. Questi pazienti non presentano iperpigmentazione, iperkaliemia e iperazotemia.
Esami di laboratorio
Alcuni sono molto aspecifici, come anemia normocitica con linfocitosi ed eosinofilia, più significative sono alcune alterazioni ematochimiche come:
- iponatriemia (<130 mEq/L), deplezione di sodio significa deplezione idrica, la quale a sua volta comporta ipovolemia e ipotensione
- iperkaliemia (>5 mEq/L) —–(responsabile di aritmie ipocinetiche) e acidosi metabolica
- ipoglicemia
- aumento dell’urea plasmatica e della creatininemia per un meccanismo prerenale che dipende dalla ipovolemia. La contrazione della volemia può essere tale da indurre una riduzione del filtrato glomerulare e quindi un aumento delle scorie azotate. Il quadro di iperazotemia prerenale regredisce non appena si provvede a reintegrare il patrimonio idrico dell’organismo.
- i livelli plasmatici di renina e ACTH sono aumentati.
Quando insufficienza corticosurrenalica è provocata da un’inadeguata produzione di ACTH ipofisaria, i livelli degli elettroliti sono nella norma, e manca la melanodermia (cosiddetto “Addison bianco”).
- L’ECG può mostrare rallentamento generalizzato del ritmo, bassi voltaggi e allungamento degli intervalli PR e QT.
Terapia sostitutiva
Il morbo di Addison è una malattia cronica ed irreversibile, quindi subordinata ad un trattamento farmacologico continuo che deve durare per tutta la vita. Dopo la diagnosi si stabilisce una adeguata terapia sostitutiva ormonale: anche se ciò non cura la causa a monte della patologia, grazie a questo intervento il paziente addisoniano potrà condurre almeno una vita praticamente normale, grazie all’eliminazione dei sintomi della carenza ormonale. E’ però molto importante, a tal fine, non saltare le dosi dei farmaci, rispettando gli orari di assunzione con scrupolo, oltre a farsi periodicamente visitare dall’endocrinologo che, tramite esami di laboratorio ed anamnesi, verificherà che la terapia sia costantemente ottimale
Terapia sostitutiva del cortisolo
In un soggetto normale la produzione giornaliera di cortisolo si aggira intorno ai 20 mg, con un ritmo circadiano sincronizzato con le fasi di sonno-veglia (massimi poco prima del risveglio e minimi nella tarda serata). In base a questi presupposti fisiologici, nel paziente affetto da morbo di Addison la terapia con sostanze glicoattive viene spesso frazionata in più dosi; esistono comunque anche glicocorticoidi a lunga durata d’azione come il desametasone (Decadron) ed il prednisone (Deltacortene) che possono essere assunti in un’unica somministrazione, di solito al momento di coricarsi. Nelle crisi addisoninane, il trattamento di emergenza eseguito dal personale sanitario si basa sull’iniezione intravenosa di glucosio, soluzione salina ed idrocortisone. Generalmente questo trattamento consente un rapido recupero. Ad ogni modo, il più delle volte la dose sostitutiva con idrocortisone (Hydrocortone, plenadren) e cortisone acetato (Cortone), viene frazionata in tre parti, di cui due vengono assunte al mattino ed una al pomeriggio (in modo da mimare il fisiologico andamento circadiano appena descritto).
Terapia sostitutiva dell’aldosterone
Non è necessaria nelle forme secondarie e si basa sull’assunzione del corticosteroide 9α-fluoroidrocortisone. Le dosi di assunzione di questo farmaco vanno adattate in base al potere mineralcorticoide del medicinale glicoattivo impiegato e ancora una volta in relazione alle condizioni del soggetto; nei mesi caldi, specie in ambienti afosi o quando si pratica un’intensa attività sportiva, ad esempio, è necessario aumentare le dosi per accentuare la ritenzione di liquidi e prevenire la disidratazione.
Terapia sostitutiva androgenica
Nelle donne con morbo di Addison viene consigliata una terapia con un debole androgeno (DHEA o deidroepiandrosterone) allo scopo di migliorare vitalità e benessere sessuale.
Trattamento della crisi surrenalica
In caso di sospetto, bisogna instaurare immediatamente la terapia. (Attenzione: durante una crisi surrenalica, un ritardo nell’avvio della terapia con corticosteroidi, in particolare se vi è ipoglicemia e ipotensione, può essere fatale). Se il paziente è in fase acuta, l’esecuzione del test di stimolazione con ormone adrenocorticotropo (ACTH) di conferma deve essere rimandata fino a quando il paziente non è migliorato. Idrocortisone 100 mg viene iniettato EV in 30 secondi e ripetuto ogni 6-8 h per le prime 24 h. Un’immediata espansione del volume intravascolare viene effettuata con 1 L di glucosio al 5% in soluzione fisiologica allo 0,9% in 1 o 2 h. Ulteriori quantità di soluzione fisiologica allo 0,9% vengono infuse fino alla correzione dell’ipotensione, della disidratazione e dell’iponatriemia. Il K sierico può diminuire bruscamente durante la fase di reidratazione, richiedendo una reintegrazione. I mineralcorticoidi non sono necessari, quando si somministrano alte dosi di idrocortisone. Quando si supera la fase acuta della malattia, si può somministrare idrocortisone 50 o 100 mg IM ogni 6 ore. Il ripristino della pressione arteriosa e il miglioramento delle condizioni generali deve verificarsi entro 1 h dalla dose iniziale di idrocortisone. Possono essere necessari farmaci inotropi, fino a quando non si rendano evidenti gli effetti dell’idrocortisone. Se il paziente è sensibilmente migliorato, durante le seconde 24 h viene solitamente somministrata una dose totale di 150 mg di idrocortisone e 75 mg vengono somministrati il 3o giorno. Successivamente, vengono somministrate quotidianamente dosi orali di mantenimento sia di idrocortisone (15-30 mg), che di fludrocortisone (0,1 mg), come descritto precedentemente. Il recupero dipende dal trattamento della causa scatenante (ad esempio un’infezione, un trauma, uno stress metabolico) e da un’adeguata terapia con idrocortisone. Per i pazienti che conservano una certa funzionalità surrenalica residua e che sviluppano una crisi surrenalica acuta in seguito a stress, il trattamento con idrocortisone è lo stesso, ma sono necessarie minori quantità di liquidi.
Prognosi, mortalità e sopravvivenza
L’aspettativa di vita per un paziente con morbo di Addison, con adeguata terapia, oscilla tra i 40 ed i 50 anni dalla diagnosi, quindi, dal momento che la malattia viene diagnosticata mediamente intorno ai 35 anni di età, nella maggioranza dei casi la malattia – se adeguatamente trattata – ha una aspettativa di vita sovrapponibile a quella della popolazione sana e solo in alcuni casi lievemente inferiore. In caso di situazioni particolarmente stressanti un morbo di Addison teoricamente controllato, può evolvere in una crisi addisoniana potenzialmente fatale.
Immagini
Leggi anche:
- Surrene: anatomia, funzioni e patologie in sintesi
- Sindrome di Cushing: immagini, sintomi, diagnosi, cura, guarigione
- Feocromocitoma e surrenectomia: sintomi e conseguenze
- Surrenectomia: tecniche e conseguenze dell’asportazione del surrene
- Iperplasia surrenale congenita: tipi, sintomi, diagnosi, cura
- Iperplasia surrenale acquisita: cause, sintomi, terapia
- Iperandrogenismo femminile: significato, cause, sintomi e terapie
- Iperandrogenismo nell’uomo: significato, cause, sintomi e terapie
- Surrene: differenza tra ormoni della parte corticale e midollare
- Deficienza di ACTH e cortisolo (ipocortisolismo secondario)
- Deficienza di CRH e cortisolo (ipocortisolismo terziario)
- Malattia di Cushing: cause, sintomi, diagnosi, terapia
- Sindrome di Achard-Thiers
- Sindrome di Waterhouse-Friderichsen: cause e sintomi
- Tumore benigno e maligno del surrene: sintomi, diagnosi, cura
- Iperaldosteronismo primario e secondario: tipi e sintomi
- Iperaldosteronismo primario (sindrome di Conn): cause, sintomi, terapia
- Iperaldosteronismo secondario: cause, sintomi e terapia
- Cos’è l’adrenalina ed a cosa serve?
- Adrenalina e “combatti o fuggi”: ecco cosa accade nel nostro corpo quando siamo terrorizzati
- Neoplasie Endocrine Multiple (MEN) tipo 1 e 2
- Testosterone basso, alto, valori normali ed interpretazione
- Ormoni estrogeni: cosa sono e quali funzioni svolgono?
- Progesterone: cos’è, a cosa serve, valori e quali funzioni ha in gravidanza?
- Quando la donna ha troppi peli dove non dovrebbero essere: irsutismo, cause, trattamenti e differenze con ipertricosi
- Cos’è una ghiandola endocrina? A che servono gli ormoni ed il sistema endocrino?
- Differenze tra ipotalamo, ipofisi, neuroipofisi e adenoipofisi
- Patologie di ipotalamo e ipofisi
- Ipotalamo: anatomia, struttura e funzioni
- Ipofisi (ghiandola pituitaria): anatomia, funzioni e ormoni secreti
- Asse ipotalamo-ipofisario: fisiologia e ormoni rilasciati
- Asse ipotalamo-ipofisi-gonade: funzionamento ed ormoni rilasciati
- Asse ipotalamo-ipofisi-testicolo: funzionamento ed ormoni rilasciati
- Asse ipotalamo-ipofisi-tiroide: funzionamento ed ormoni rilasciati
- Differenze tra adrenalina e noradrenalina
- Apparato urinario: anatomia e fisiologia [SCHEMA]
- Rene: anatomia, funzioni e patologie in sintesi
- Differenza tra surrene e rene
- Dopammina: cos’è ed a che serve?
- Neurotrasmettitori: cosa sono ed a che servono
- Si può vivere senza reni? Conseguenze della nefrectomia
- Dopammina: biosintesi, rilascio nello spazio sinaptico e degradazione
- Sistema dopamminergico: i circuti nervosi della dopammina
- Colesterolo: cos’è ed a cosa serve?
- Adrenalina ed epinefrina sono la stessa cosa?
- Ormone della crescita (GH) a che serve e da cosa è prodotto?
- Serotonina e triptofano: cosa sono e in quali cibi trovarli
- Glutamina: a che serve, quando assumerlo, dosi ed effetti collaterali
- Creatinina alta o bassa: cos’è, cosa indica e come si corregge
- Rene: anatomia, funzioni e patologie in sintesi
- Sistema nervoso: com’è fatto, a che serve e come funziona
- Sistema nervoso simpatico: funzioni
- Sistema nervoso parasimpatico: funzioni
Lo Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o seguici su Twitter, su Instagram o su Pinterest, grazie!
