
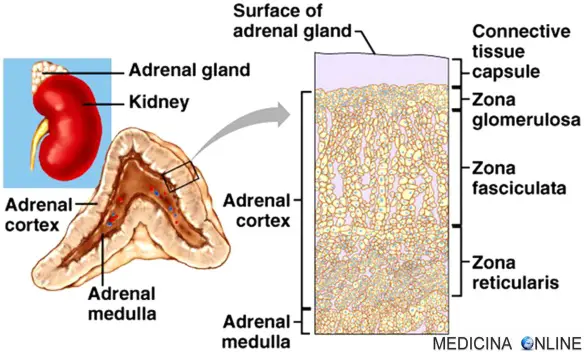
 L’iperplasia surrenale congenita (o “CAH“) è una patologia ereditaria che colpisce le ghiandole surrenali, poste sopra i reni. È una malattia autosomica recessiva, caratterizzata da disordini nella biosintesi degli ormoni steroidei nella corteccia surrenale, a causa di una mutazione in uno dei 5 enzimi coinvolti nel processo di sintesi. Ricordiamo che la corteccia surrenalica è la parte più esterna del surrene (quella più interna è la midollare che produce catecolamine come l’adrenalina) e secerne gli ormoni steroidei (corticosteroidi ed ormoni sessuali). E’ divisa in tre zone:
L’iperplasia surrenale congenita (o “CAH“) è una patologia ereditaria che colpisce le ghiandole surrenali, poste sopra i reni. È una malattia autosomica recessiva, caratterizzata da disordini nella biosintesi degli ormoni steroidei nella corteccia surrenale, a causa di una mutazione in uno dei 5 enzimi coinvolti nel processo di sintesi. Ricordiamo che la corteccia surrenalica è la parte più esterna del surrene (quella più interna è la midollare che produce catecolamine come l’adrenalina) e secerne gli ormoni steroidei (corticosteroidi ed ormoni sessuali). E’ divisa in tre zone:
- zona glomerulare: è la parte più esterna della corticale del surrene e produce mineralcorticoidi come l’aldosterone che controlla i livelli di elettroliti e la quantità di acqua presenti nel sangue;
- zona fascicolata: è la parte centrale della corticale del surrene e produce glucocorticoidi come il cortisolo che controlla il metabolismo di carboidrati, lipidi e proteine;
- zona reticolata: è la parte più interna della corticale del surrene e produce soprattutto androgeni (ormoni sessuali maschili come il testosterone) e piccole quantità di estrogeni (ormoni sessuali femminili).
Gli ormoni prodotti nella parte corticale del surrene sono coinvolti in una varietà di meccanismi fisiologici, inclusi quelli che regolano l’infiammazione, il sistema immunitario, il metabolismo dei carboidrati, delle proteine, il livello di elettroliti nel sangue, la sessualità e la riproduzione. La mutazione enzimatica comporta una alterata produzione di ormoni glucocorticoidi e mineralcorticoidi, con conseguente aumento di androgeni.
Iperplasia congenita e acquisita
Ricordiamo che l’alterazione della corteccia surrenale tipica nell’iperplasia surrenalica congenita, può essere dovuta anche a
- iperplasia acquisita;
- adenomi;
- adenocarcinomi.
Cause
Nel 90% dei casi, la malattia dipende dal deficit della 21-idrossilasi derivato dai surreni, questo comporta una disfunzione della ghiandola surrenale. L’enzima 21-idrossilasi, appartiene alla famiglia dei citocromi P450, e converte il 17-OH-Progesterone in 11-Deossicortisolo, precursore del cortisolo. Come tutti i citocromi, l’enzima accetta elettroni dal citocromo P450 reduttasi NADH-dipendente, riducendo l’ossigeno molecolare e idrossilando il substrato. La CYP-21 umana è fatta da 494 aminoacidi, con un peso molecolare di 55 KDa. Il deficit da questo enzima comporta la deviazione della sintesi ormonale verso quei metaboliti che non richiedono tale enzima per la loro sintesi, come testosterone, androstenedione, estrogeni, estrone ed estradiolo. La mancanza di CYP-21 determina inoltre accumulo dei composti intermedi, come il 17-idrossi-progesterone. L’ipofisi, che funziona come una centralina di regolazione ormonale, registra l’assenza in circolo di cortisolo e produce ACTH (ormone adrenocorticotropo), la quale stimola le ghiandole surrenali a cominciare le tappe di produzione del cortisolo. Un eccesso di ACTH porterà accumulo di 17-idrossi-progesterone, dal quale derivano gli androgeni (ormoni sessuali maschili). Ulteriori difetti enzimatici possono essere la compromissione della sintesi di aldosterone, utile al mantenimento dell’omeostasi del sodio, con conseguente perdita di sale, squilibrio idroelettrolitico con ipovolemia e shock. L’eccesso di androgeni in entrambi i sessi si manifesta con accelerazione della maturità ossea, acne, irsutismo, fino ad avere virilizzazione.
Trasmissione
L’iperplasia surrenale congenita è una malattia autosomica recessiva, per approfondire, leggi: Differenza tra autosomica dominante e recessiva con esempi
Sintomi e segni
In base all’attività enzimatica di CYP-21, e alle caratteristiche fenotipiche delle persone affette, classifichiamo le forme di questa iperplasia in una forma classica ed in una non classica.
Forma classica
La cosiddetta forma classica si manifesta in epoca neonatale o nelle prime fasi dell’infanzia (in Italia, 1:16000 nati). Può essere di due tipi:
- virilizzazione semplice: In questi pazienti non si producono sufficienti quantità di cortisolo, ma si hanno adeguate quantità di aldosterone che consentono di avere un corretto bilancio elettrolitico;
- con perdita di sale: in questo caso si ha deficit enzimatico totale, e non vengono prodotti né cortisolo né aldosterone (insufficienza surrenalica). Nei pazienti con deficit di cortisolo si ha un peggioramento della funzione cardiaca, con scarsa risposta vascolare alle catecolamine e una ridotta velocità di filtrazione glomerulare. L’assenza di entrambi gli ormoni si manifesta in maschi e femmine con vomito, diarrea, perdita di peso, disidratazione, shock.
Forma non classica
La cosiddetta non classica può essere asintomatica o associata a pochi segni di iperandrogenismo, e in genere si manifesta tardivamente. Frequenze: (ebrei ashkenazi, 1:27; ispanici, 1:53; slavi 1:63; italiani, 1:333; caucasici, 1:1000). Questo suggerisce che più dell’1% della popolazione è eterozigote, e quindi portatrice dell’allele NC. La forma non classica può essere di 2 tipi:
- a insorgenza tardiva: In questi pazienti si verificano manifestazioni dovute a iperandrogenismo, come comparsa precoce di peluria pubica ed ascellare, modeste accelerazioni della crescita e della maturazione scheletrica, acne, irsutismo, PCOS (sindrome dell’ovaio policistico). Nei maschi si può avere azospermia, oligospermia, infertilità;
- criptica: Questa forma è completamente asintomatica, e prevalgono i segni clinici legati ad una condizione di iperandrogenismo moderato-lieve.
L’iperandrogenismo sia nella forma classica che in quella non classica porta allo sviluppo della PCOS, con conseguente oligomenorrea, amenorrea, soprattutto in adolescenza. Inoltre, sebbene la fertilità possa essere ridotta, anche se la produzione di androgeni non viene soppressa, una donna può concepire e portare a termine una gravidanza con successo. L’infertilità si ha solo nel 13% delle donne con deficit non classico di 21-idrossilasi, Si mostrano iperplasia del clitoride o del pene, bassa altezza a crescita terminata, acne grave, ipertricosi, disturbi mestruali, infertilità.
Leggi anche:
- Virilizzazione (mascolinizzazione): tipi, cause, sintomi, diagnosi e terapie
- Irsutismo: cause, sintomi, diagnosi e trattamenti
Diagnosi
Sospettata nei neonati con grave deficit dell’accrescimento ponderale, e in caso di ipertrofia del clitoride. Si utilizzano esami ematochimici (analisi del sangue) o preventivamente durante lo screening neonatale. Nella forma classica:
- nelle femmine la diagnosi si effettua alla nascita, per la presenza di ambiguità genitali;
- nel maschio la diagnosi si fa per lo sviluppo di pubertà precoce tra i due e i quattro anni.
Nella forma non classica:
- nella donna prevalgono i segni clinici legati a iperandrogenismo moderato-lieve; irsutismo, oligomenorrea e acne sono i sintomi frequenti.
Trattamento
Il trattamento farmacologico consiste nel correggere il deficit di glucorticoidi e mineralcorticoidi. I glucocorticoidi sopprimono l’eccessiva secrezione di ACTH dall’ipotalamo, riducendo così i livelli ormonali di steroidi sessuali surrenalici, evitando virilizzazione genitale e permettendo un normale accrescimento staturale e puberale. Nell’adulto il trattamento deve continuare per evitare l’iperandrogenismo, per mantenere il normale ciclo mestruale e la fertilità nelle donne. Il glucocorticoide più usato è l’idrocortisone. Il trattamento chirurgico invece consiste nella genito plastica femminilizzante, per ricreare l’anatomia dei genitali esterni per dare alla paziente con pseudoermafroditismo una normalità estetica e funzionale.
Leggi anche:
- Iperplasia surrenale acquisita: cause, sintomi, terapia
- Surrene: anatomia, funzioni e patologie in sintesi
- Tumore benigno e maligno del surrene: sintomi, diagnosi, cura
- Morbo di Addison: sintomi, immagini, terapia, mortalità, aspettative di vita
- Sindrome di Cushing: immagini, sintomi, diagnosi, cura, guarigione
- Feocromocitoma e surrenectomia: sintomi e conseguenze
- Surrenectomia: tecniche e conseguenze dell’asportazione del surrene
- Iperandrogenismo femminile: significato, cause, sintomi e terapie
- Iperandrogenismo nell’uomo: significato, cause, sintomi e terapie
- Intersessualità: cause, tipi, sintomi, terapia
- Ermafrodita, ermafroditismo, pseudoermafroditismo maschile e femminile, cosa significa?
- Androgino e androginia: etimologia, sessualità, genitali, mito di Platone
- Differenza tra ermafrodita e androgino
- Sindrome di Reifenstein (parziale insensibilità agli androgeni): cause, sintomi, diagnosi, terapia, complicanze
- Sindrome di Swyer (disgenesia gonadica pura) e gonadi a striscia
- Sindrome di Perrault: cause, sintomi, diagnosi, terapia, prognosi
- Sindrome dell’ovaio resistente (sindrome di Savage)
- Surrene: differenza tra ormoni della parte corticale e midollare
- Deficienza di ACTH e cortisolo (ipocortisolismo secondario)
- Deficienza di CRH e cortisolo (ipocortisolismo terziario)
- Malattia di Cushing: cause, sintomi, diagnosi, terapia
- Sindrome di Achard-Thiers
- Sindrome di Waterhouse-Friderichsen: cause e sintomi
- Iperaldosteronismo primario e secondario: tipi e sintomi
- Iperaldosteronismo primario (sindrome di Conn): cause, sintomi, terapia
- Iperaldosteronismo secondario: cause, sintomi e terapia
- Asse ipotalamo-ipofisi-surrene: funzionamento ed ormoni rilasciati
- Cos’è l’adrenalina ed a cosa serve?
- Adrenalina e “combatti o fuggi”: ecco cosa accade nel nostro corpo quando siamo terrorizzati
- Neoplasie Endocrine Multiple (MEN) tipo 1 e 2
- Testosterone basso, alto, valori normali ed interpretazione
- Ormoni estrogeni: cosa sono e quali funzioni svolgono?
- Progesterone: cos’è, a cosa serve, valori e quali funzioni ha in gravidanza?
- Quando la donna ha troppi peli dove non dovrebbero essere: irsutismo, cause, trattamenti e differenze con ipertricosi
- Cos’è una ghiandola endocrina? A che servono gli ormoni ed il sistema endocrino?
- Differenze tra ipotalamo, ipofisi, neuroipofisi e adenoipofisi
- Patologie di ipotalamo e ipofisi
- Ipotalamo: anatomia, struttura e funzioni
- Ipofisi (ghiandola pituitaria): anatomia, funzioni e ormoni secreti
- Asse ipotalamo-ipofisario: fisiologia e ormoni rilasciati
- Asse ipotalamo-ipofisi-gonade: funzionamento ed ormoni rilasciati
- Asse ipotalamo-ipofisi-testicolo: funzionamento ed ormoni rilasciati
- Asse ipotalamo-ipofisi-tiroide: funzionamento ed ormoni rilasciati
- Differenze tra adrenalina e noradrenalina
- Apparato urinario: anatomia e fisiologia [SCHEMA]
- Rene: anatomia, funzioni e patologie in sintesi
- Differenza tra surrene e rene
- Dopammina: cos’è ed a che serve?
- Neurotrasmettitori: cosa sono ed a che servono
- Si può vivere senza reni? Conseguenze della nefrectomia
- Dopammina: biosintesi, rilascio nello spazio sinaptico e degradazione
- Sistema dopamminergico: i circuti nervosi della dopammina
- Colesterolo: cos’è ed a cosa serve?
- Adrenalina ed epinefrina sono la stessa cosa?
- Ormone della crescita (GH) a che serve e da cosa è prodotto?
- Serotonina e triptofano: cosa sono e in quali cibi trovarli
- Glutamina: a che serve, quando assumerlo, dosi ed effetti collaterali
- Creatinina alta o bassa: cos’è, cosa indica e come si corregge
- Rene: anatomia, funzioni e patologie in sintesi
- Sistema nervoso: com’è fatto, a che serve e come funziona
- Sistema nervoso simpatico: funzioni
- Sistema nervoso parasimpatico: funzioni
Lo Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o seguici su Twitter, su Instagram o su Pinterest, grazie!
