
 La sindrome di Gilbert è una patologia benigna del fegato che si manifesta con iperbilirubinemia, cioè un aumento della bilirubina nel sangue, spesso nel secondo decennio di vita. Ne è affetto circa il 7-8% della popolazione adulta ed è a carattere ereditario, trasmessa con modalità sia autosomico recessiva che dominante.
La sindrome di Gilbert è una patologia benigna del fegato che si manifesta con iperbilirubinemia, cioè un aumento della bilirubina nel sangue, spesso nel secondo decennio di vita. Ne è affetto circa il 7-8% della popolazione adulta ed è a carattere ereditario, trasmessa con modalità sia autosomico recessiva che dominante.
Cause
Nella sindrome di Gilbert l’iperbilirubinemia è causata da una ridotta attività della glucuronosiltransferasi (UGT), fondamentale per la coniugazione della bilirubina, che diviene così idrosolubile rendendo possibile l’escrezione biliare. Nei pazienti affetti da sindrome di Gilbert la bilirubina non viene adeguatamente escreta e la sua concentrazione ematica aumenta. In questa malattia è, dunque, presente un deficit parziale dell’attività dell’enzima UDP-glucoronil transferasi, in particolare, della isoforma UGT1A1. L’entità di tale deficit, compreso tra il 20 e il 70% del valore normale, determina la diversa espressione clinica e gravità sintomatologica (ittero) della malattia. Ciò comporta livelli di bilirubina generalmente di poco sopra la norma che possono aumentare momentaneamente in condizioni quali: digiuno, semidigiuno, ingestione di alcol, stress, febbre, infezioni, aumento dell’attività fisica. Se la quantità di bilirubina è elevata (maggiore di 2.5 mg/dl), si può manifestare l’ittero (colorazione gialla) della pelle e delle sclere (la parte bianca degli occhi). Nel paziente con sindrome di Gilbert anche la produzione di insulina è leggermente superiore alla norma e non costante nell’arco della giornata. Ciò può determinare una carenza di nutrimento alle cellule nervose, contribuendo così a una depressione del tono neuro-umorale.
Trasmissione
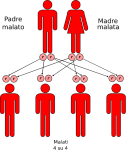
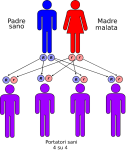
La trasmissione è autosomica recessiva, il che significa che l’allele alterato deve essere presente in coppia (omozigosi), cioè sono necessarie due copie dell’allele difettoso per far sì che la malattia si esprima, a prescindere dal sesso. Non basta un solo genitore portatore sano o malato, bensì entrambi i genitori devono essere portatori sani o malati. Il fenotipo quindi si esprime quando nel genotipo dell’individuo sono presenti entrambi gli alleli responsabili, fatto che spiega l’alta probabilità di sviluppare malattie genetiche in caso di incesto. Quindi:
- un individuo che possegga entrambi gli alleli alterati: è portatore ed è malato;
- un individuo che possegga solo un allele alterato: è portatore ma è sano;
- un individuo che non possegga nessun allele alterato: NON è portatore ed è sano.
Essere portatore sano vuol dire quindi NON avere la patologia ma possedere nel proprio genotipo un allele mutato, che può essere trasmesso alle generazioni successive.
Dalla combinazione delle possibili condizioni di genitori sani, malati e portatori sani, deriva la distribuzione probabilità che la malattia sia trasmessa ai figli:
- genitori malato-malato: la probabilità che il figlio/a nasca malato è del 100%;
- genitori sano-malato: la probabilità che il figlio/a nasca portatore sano è del 100%;
- genitori malato-portatore sano: la probabilità che il figlio/a nasca malato è del 50% e del 50% che nasca portatore sano;
- genitori sano-portatore sano: la probabilità che il figlio/a nasca sano è del 50% e del 50% che nasca portatore sano;
- genitori portatore-portatore: la probabilità che il figlio/a nasca portatore sano è del 50% mentre è del 25% che nasca sano o malato.
Se nessuno dei genitori ha un allele mutato, non c’è ovviamente alcuna trasmissione autosomica recessiva ed i figli saranno tutti sani e NON portatori dell’allele mutato.
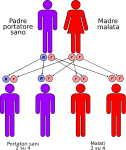
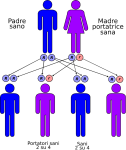
Nell’immagine che segue, è raffigurata la tipica situazione in cui entrambi i genitori sono sani ma portatori dell’allele mutato:
- un figlio su quattro avrà entrambi gli alleli alterati e sarà malato ed ovviamente portatore;
- due figli su quattro avranno un allele normale ed uno alterato e saranno sani ma anche portatori;
- un figlio su quattro avrà entrambi gli alleli normali e sarà sano e NON portatore.

Le altre quattro situazioni possibili sono raffigurate nelle seguenti immagini:
Sintomi
Un terzo dei pazienti affetti dalla sindrome è completamente asintomatico, mentre i rimanenti due terzi accusano disturbi aspecifici quali dolori addominali, affaticabilità, cefalee e malessere. Nel periodo manifestante della sindrome, possono verificarsi momenti di depressione o semplice debolezza.
Diagnosi e valori
In genere si può parlare di sindrome di Gilbert quando si è in presenza di un aumento sostanziale della “bilirubina indiretta” (valori normali inferiori a 1 mg/100 ml) e di un lieve o inesistente aumento della “bilirubina diretta” (valori normali inferiori a 0,5 mg/100 ml). Gli esami si effettuano a digiuno. Importante è la diagnosi differenziale in caso di ittero o subittero con altre malattie epatiche che si manifestano con iperbilirubinemia. Ciò è possibile con semplici esami di laboratorio in quanto la sindrome di Gilbert non comporta danni funzionali al fegato, che quindi non presenterà ALT, AST, GGT alterati, la sintesi proteica epatica (valutabile mediante PT, PTT, albuminemia, protidemia..) sarà nella norma, l’emocromo non presenterà eritrocitopenia (da possibile emolisi). Un metodo economico per diagnosticare il Gilbert consiste nel test del digiuno. Se dopo una giornata di ridotto apporto calorico (una fetta di pane, una fettina di carne e un po’ di frutta) la bilirubina sale, mentre tutti gli altri parametri restano nei range di riferimento, è suggestivo di Gilbert. Sono comunque disponibili test genetici per fare diagnosi molecolare di Gilbert.
Invalidità
I pazienti affetti da sindrome di Gilbert attualmente non ha esenzione ticket e gradi di invalidità.
Trattamento
Non esiste alcun tipo di terapia in quanto si tratta di un’alterazione benigna che solo in basse percentuali può influire sulla vita dei soggetti. Nonostante questo è da segnalare che la somministrazione di barbiturici può attenuare la sintomatologia itterica, aumentando l’attività dell’UDP glucoronil transferasi. Tuttavia, la sindrome di Gilbert riduce la capacità del fegato di detossificare determinati farmaci. Per esempio, la sindrome di Gilbert è associata a forte diarrea e neutropenia in pazienti che assumono irinotecano, che viene metabolizzato da UGT1A1 (Glucuronosiltransferasi).
Leggi anche:
- Bilirubina diretta e indiretta: ittero, significato, patologie collegate
- Ittero emolitico, colestatico, ostruttivo, neonatale: significato, occhi, cura
- Pelle gialla: differenza tra ittero e carotenodermia
- Anemia emolitica: farmaci, bilirubina, ittero, diagnosi di laboratorio
- Insufficienza epatica lieve, acuta e cronica: dieta e rischio di morte
- Bile: dove si trova, a che serve e da cosa è composta?
- Cirrosi epatica e fegato: sintomi, dieta, diagnosi, terapia e prevenzione
- Funzionalità epatica; cos’è, cosa indica e come si misura
- Foetor hepaticus: causa, odore, definizione, significato
- Epatiti croniche: cosa sono, sintomi, diagnosi e cura
- Transaminasi alte, basse, cosa sono, cosa indicano e come si curano
- Vena porta e sistema portale: anatomia e funzioni della circolazione epatica
- Legatura/sclerosi delle varici esofagee: perché si esegue, quali sono i rischi?
- Elastografia epatica (FibroScan) per cirrosi: valori, preparazione all’esame, risultati
- Calcolosi colecisti: sintomi, dieta e terapie dei calcoli biliari
- Colecistite acuta e cronica litiasica e alitiasica: cause, terapia, dieta e rimedi naturali
- Dipendenza da alcol: come fare per smettere di bere alcolici e superalcolici
- L’alcol è una droga?
- Dove si trova il fegato ed a che serve?
- Differenza tra cirrosi e fibrosi
- Fegato ed epatociti: anatomia, funzioni e patologie in sintesi
- L’esofagogastroduodenoscopia: cos’è, preparazione, è dolorosa o pericolosa?
- Colangiopancreatografia retrograda (ERCP): cos’è, preparazione, è dolorosa o pericolosa?
- Fegato ed epatociti: anatomia, funzioni e patologie in sintesi
- Cistifellea: cos’è, a cosa serve e dove si trova
- Dotto epatico comune, cistico e coledoco: anatomia del sistema biliare
- Duodeno: anatomia e funzioni in sintesi
- Pancreas: anatomia e funzioni in sintesi
- Cistifellea: cos’è, a cosa serve e dove si trova
- Si può vivere senza cistifellea?
- Riconoscere i differenti tipi di vomito a seconda del colore
- Vomito: le cause più frequenti
- Vomito: rimedi naturali e cure farmacologiche (farmaci anti-emetici)
- Sindrome post-colecistectomia: conseguenze dell’asportazione della cistifellea
- Differenza tra intestino tenue e crasso
- Mal di pancia e di stomaco: da cosa può dipendere e quali sono le cure
- Mal di pancia forte: quando chiamare il medico?
- Differenza tra ulcera gastrica, duodenale, peptica ed esofagea
- Ulcera peptica: complicanze, cura, dieta, quando è pericolosa
- Ecografia renale: esecuzione, indicazioni, costo
- Ecografia alla vescica vuota o piena: preparazione, costo, svolgimento
- Feci dalla bocca: il vomito fecaloide
- Incontinenza fecale: dieta, Alzheimer, parto, cause e terapie
- Il mio alito odora di feci: cause, quando è pericoloso e rimedi
- Morbo di Crohn: cos’è, cause scatenanti, sintomi, cure e dieta
- Differenze tra ileo meccanico ed ileo paralitico: cause, sintomi e trattamenti
- Colite ulcerosa: cause, diagnosi, cura, dieta, cosa mangiare, rimedi
- Differenza tra colon irritabile, colite e colite spastica: sono la stessa cosa?
Lo Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o seguici su Twitter, su Instagram o su Pinterest, grazie!
