
 La malattia di Wilson (in passato chiamata “morbo di Wilson”) è una patologia ereditaria autosomica recessiva caratterizzata dalla ridotta eliminazione del rame nella bile da parte del fegato. Tale difetto comporta un accumulo di rame nel fegato e in altri organi quali il sistema nervoso centrale, la cornea (dove si può formare l’anello di Kayser-Fleischer, vedi paragrafo specifico), il rene, le ossa. L’accumulo di rame danneggia consistentemente questi organi ed è responsabile della comparsa di sintomi e segni clinici. La malattia prende il nome dal neurologo inglese Samuel Alexander Kinnier Wilson (1878-1937), che per primo la descrisse nel 1912.
La malattia di Wilson (in passato chiamata “morbo di Wilson”) è una patologia ereditaria autosomica recessiva caratterizzata dalla ridotta eliminazione del rame nella bile da parte del fegato. Tale difetto comporta un accumulo di rame nel fegato e in altri organi quali il sistema nervoso centrale, la cornea (dove si può formare l’anello di Kayser-Fleischer, vedi paragrafo specifico), il rene, le ossa. L’accumulo di rame danneggia consistentemente questi organi ed è responsabile della comparsa di sintomi e segni clinici. La malattia prende il nome dal neurologo inglese Samuel Alexander Kinnier Wilson (1878-1937), che per primo la descrisse nel 1912.
Sinonimi
La malattia di Wilson in passato veniva chiamata “morbo di Wilson“, espressione che ormai è divenuta rara. Sinonimo di malattia di Wilson è “degenerazione epatolenticolare“. In inglese viene chiamata Wilson’s disease. La malattia di Wilson non deve essere confusa con la sindrome di Wilson (in inglese Wilson’s temperature syndrome o Wilson’s thyroid syndrome).
Diffusione
E’ una malattia che colpisce tra 1 ed 8 individui ogni 100 mila persone.
Cause
La patologia è dovuta ad una mutazione nella proteina della malattia di Wilson (gene ATP7B) mappato sul cromosoma 13. Una singola copia anomala del gene è presente in una persona su cento, senza determinare alcun sintomo, essendo la patologia causata da un gene recessivo (si tratta di portatori sani della malattia). Se un individuo eredita il gene da entrambi i genitori, è a rischio di manifestare la malattia.
Trasmissione
La trasmissione è autosomica recessiva, il che significa che l’allele alterato deve essere presente in coppia (omozigosi), cioè sono necessarie due copie dell’allele difettoso per far sì che la malattia si esprima, a prescindere dal sesso. Non basta un solo genitore portatore sano o malato, bensì entrambi i genitori devono essere portatori sani o malati. Il fenotipo quindi si esprime quando nel genotipo dell’individuo sono presenti entrambi gli alleli responsabili, fatto che spiega l’alta probabilità di sviluppare malattie genetiche in caso di incesto. Quindi:
- un individuo che possegga entrambi gli alleli alterati: è portatore ed è malato;
- un individuo che possegga solo un allele alterato: è portatore ma è sano;
- un individuo che non possegga nessun allele alterato: NON è portatore ed è sano.
Essere portatore sano vuol dire quindi NON avere la patologia ma possedere nel proprio genotipo un allele mutato, che può essere trasmesso alle generazioni successive.
Dalla combinazione delle possibili condizioni di genitori sani, malati e portatori sani, deriva la distribuzione probabilità che la malattia sia trasmessa ai figli:
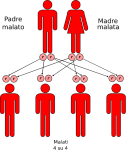
- genitori malato-malato: la probabilità che il figlio/a nasca malato è del 100%;
- genitori sano-malato: la probabilità che il figlio/a nasca portatore sano è del 100%;
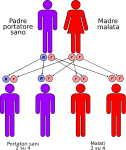
- genitori malato-portatore sano: la probabilità che il figlio/a nasca malato è del 50% e del 50% che nasca portatore sano;
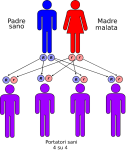
- genitori sano-portatore sano: la probabilità che il figlio/a nasca sano è del 50% e del 50% che nasca portatore sano;
- genitori portatore-portatore: la probabilità che il figlio/a nasca portatore sano è del 50% mentre è del 25% che nasca sano o malato.
Se nessuno dei genitori ha un allele mutato, non c’è ovviamente alcuna trasmissione autosomica recessiva ed i figli saranno tutti sani e NON portatori dell’allele mutato.
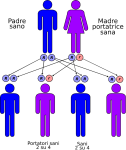
Nell’immagine che segue, è raffigurata la tipica situazione in cui entrambi i genitori sono sani ma portatori dell’allele mutato:
- un figlio su quattro avrà entrambi gli alleli alterati e sarà malato ed ovviamente portatore;
- due figli su quattro avranno un allele normale ed uno alterato e saranno sani ma anche portatori;
- un figlio su quattro avrà entrambi gli alleli normali e sarà sano e NON portatore.

Le altre quattro situazioni possibili sono raffigurate nelle seguenti immagini:
Sintomi e segni
Nei bambini e negli adolescenti la malattia di Wilson si manifesta più frequentemente con una patologia del fegato, mentre nei giovani e negli adulti si può manifestare anche con problemi neurologici. In alcuni casi la malattia di Wilson può esordire con disturbi del comportamento e della sfera psichica che possono simulare una patologia psichiatrica. Il tempestivo riconoscimento della malattia è reso difficoltoso dal fatto che la malattia epatica spesso decorre in modo asintomatico e quando la malattia di Wilson si presenta in modo sintomatico nessuno dei segni clinici di presentazione è esclusivo. La malattia epatica può essere diagnosticata in seguito al riscontro di alterazioni delle transaminasi e/o per un deficit degli indici di sintesi epatica (albumina e fattori della coagulazione). Talora la malattia può presentarsi con una complicanza della cirrosi (ascite, emorragia digestiva), in rari casi come un’epatite fulminante caratterizzata da insufficienza epatica grave associata ad anemia emolitica.
Il danno del sistema nervoso si può esprimere con tremori, incapacità a svolgere attività che richiedono una buona coordinazione tra la vista e le mani, alterazioni del tono muscolare, difficoltà ad articolare le parole, movimenti involontari. Anche i sintomi psichici non hanno specificità assoluta: frequentemente prendono la forma di un comportamento incongruo, irritabilità, depressione, allucinazioni ed idee deliranti.
Anello di Kayser-Fleischer
Un segno tipico della malattia di Wilson, è l’anello di Kayser-Fleischer: così viene chiamata una formazione circolare giallo-bruna-verdastra che insorge nella periferia della cornea, a livello della membrana di Descemet, in seguito al deposito ed all’accumulo di rame nel tessuto della cornea (vedi immagine in basso). Tale segno deve il suo nome a i due medici tedeschi he per primi lo descrissero nel 1902 e 1903, Bernhard Kayser e Bruno Fleischer. In inglese viene chiamato Kayser–Fleischer ring (KF rings). In italiano a volte si usa l’acronimo “anello KF” o “AKF“.

Sintomi e segni neurologici e psichiatrici
Circa la metà dei pazienti con malattia di Wilson presenta problemi neurologici o psichiatrici. La maggior parte dei pazienti inizialmente manifesta un deterioramento lieve delle capacità cognitive, assieme a cambiamenti comportamentali. In seguito giungono sintomi neurologici specifici, spesso sotto forma di parkinsonismo (aumentata rigidità e rallentamento dei comuni movimenti), con o senza un tipico tremore alle mani, espressioni facciali mascherate, difetti nell’articolazione del linguaggio, atassia (assenza di coordinazione) o distonia (improvvisi ed incontrollati movimenti di parti del corpo). I sintomi più comuni nel morbo di Wilson sono convulsioni ed emicrania. Problemi psichiatrici causati dalla malattia di Wilson possono includere cambi comportamentali quali la depressione, l’ansia e la psicosi. I sintomi psichiatrici sono comunemente osservati insieme con i sintomi neurologici, raramente si manifestano da soli. Questi sintomi spesso non sono ben definiti e si possono attribuire ad altre cause. Per questo la diagnosi del morbo di Wilson è raramente raggiunta quando solo i sintomi psichiatrici sono presenti.
Diagnosi
La malattia di Wilson può essere sospettata sulla base di uno qualsiasi dei sintomi di cui sopra oppure quando a un parente stretto ne è stato trovato affetto. La maggior parte dei pazienti presenta test di funzionalità epatica anomali, come: elevate transaminasi, alanina transaminasi e bilirubina. Se il danno epatico è significativo, i livelli di albumina possono essere bassi a causa di una incapacità delle cellule epatiche danneggiate di produrre questa proteina. Allo stesso modo, il tempo di protrombina (un test di coagulazione del sangue) può essere prolungato, in quanto il fegato non è in grado di produrre proteine note come fattori della coagulazione. I livelli di fosfatasi alcalina sono relativamente bassi nei soggetti ammalati e questo è legato all’insufficienza epatica acuta. Se sono presenti sintomi neurologici, la risonanza magnetica del cervello viene di solito eseguita; essa può mostrare anche il “volto del panda gigante” caratteristico della malattia. Non esiste un test completamente affidabile per la diagnosi del morbo di Wilson, ma i livelli di ceruloplasmina e rame plasmatici, nonché della quantità di rame escreto nelle urine durante un periodo di 24 ore, sono utilizzati per capire la quantità di rame presente nel corpo. La prova standard ideale per la diagnosi è comunque la biopsia epatica.
Leggi anche:
- Funzionalità epatica; cos’è, cosa indica e come si misura
- Transaminasi alte, basse, cosa sono, cosa indicano e come si curano
- Albumina ed albuminemia alta o bassa: cause, valori e terapie
- Tempo di protrombina (PT): valori normali, INR alto, basso, cosa fare
Terapia
La malattia di Wilson, una volta diagnosticata, può essere efficacemente controllata attraverso una opportuna terapia farmacologica. Il tipo di terapia dovrà essere stabilito dal centro specialistico in base all’età del paziente, al quadro clinico presente all’esordio, al comportamento clinico-laboratoristico del paziente in corso di terapia.
Dieta
La terapia è basata anche su una dieta a basso contenuto di rame, a tal proposito vi invitiamo a leggere: Classifica dei cibi con maggiori e minori quantità di rame esistenti
Prognosi, aspettativa di vita, mortalità
La malattia di Wilson, quando è adeguatamente trattata, ha una prognosi eccellente con una aspettativa di vita che coincide con quella della popolazione generale. Il trattamento deve però essere proseguito per tutta la vita SENZA ECCEZIONI. Il soggetto affetto da malattia di Wilson, soprattutto se diagnosticato in età pediatrica e in tutti i casi in cui la malattia è stata diagnosticata precocemente e non ha avuto il tempo di danneggiare l’organismo, può svolgere una vita normale senza alcuna limitazione rispetto alla popolazione sana. Se, al contrario, la malattia è stata diagnosticata tardi e/o se non si è seguito correttamente l’iter terapeutico, i danni epatici e cerebrali potrebbero essere irreparabili (quindi permanere per tutta la vita) o solo in parte curabili: ricordate che prima si inizia la terapia e migliori saranno la prognosi e la qualità della vita. Nei casi più gravi, l’unica vera soluzione per una prognosi migliore per il paziente rimane il trapianto di fegato.
Leggi anche:
- Insufficienza epatica lieve, acuta e cronica: dieta e rischio di morte
- Cirrosi epatica e fegato: sintomi, dieta, diagnosi, terapia e prevenzione
- Foetor hepaticus: causa, odore, definizione, significato
- Epatiti croniche: cosa sono, sintomi, diagnosi e cura
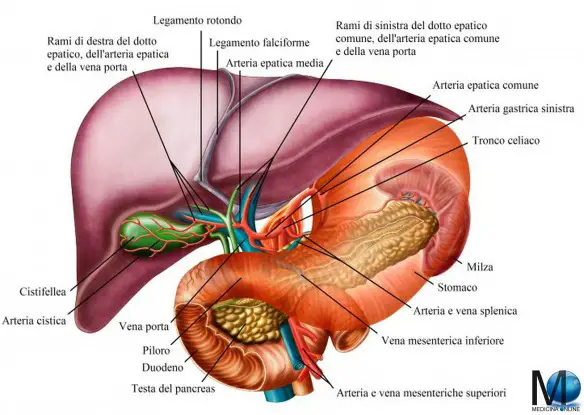
- Vena porta e sistema portale: anatomia e funzioni della circolazione epatica
- Legatura/sclerosi delle varici esofagee: perché si esegue, quali sono i rischi?
- Elastografia epatica (FibroScan) per cirrosi: valori, preparazione all’esame, risultati
- Calcolosi colecisti: sintomi, dieta e terapie dei calcoli biliari
- Colecistite acuta e cronica litiasica e alitiasica: cause, terapia, dieta e rimedi naturali
- Dipendenza da alcol: come fare per smettere di bere alcolici e superalcolici
- L’alcol è una droga?
- Dove si trova il fegato ed a che serve?
- Differenza tra cirrosi e fibrosi
- Bile: dove si trova, a che serve e da cosa è composta?
- Bilirubina diretta e indiretta: ittero, significato, patologie collegate
- Ittero emolitico, colestatico, ostruttivo, neonatale: significato, occhi, cura
- Pelle gialla: differenza tra ittero e carotenodermia
- Anemia emolitica: farmaci, bilirubina, ittero, diagnosi di laboratorio
- Fegato ed epatociti: anatomia, funzioni e patologie in sintesi
- L’esofagogastroduodenoscopia: cos’è, preparazione, è dolorosa o pericolosa?
- Colangiopancreatografia retrograda (ERCP): cos’è, preparazione, è dolorosa o pericolosa?
- Fegato ed epatociti: anatomia, funzioni e patologie in sintesi
- Cistifellea: cos’è, a cosa serve e dove si trova
- Dotto epatico comune, cistico e coledoco: anatomia del sistema biliare
- Duodeno: anatomia e funzioni in sintesi
- Pancreas: anatomia e funzioni in sintesi
- Cistifellea: cos’è, a cosa serve e dove si trova
- Si può vivere senza cistifellea?
- Riconoscere i differenti tipi di vomito a seconda del colore
- Vomito: le cause più frequenti
- Vomito: rimedi naturali e cure farmacologiche (farmaci anti-emetici)
- Sindrome post-colecistectomia: conseguenze dell’asportazione della cistifellea
- Differenza tra intestino tenue e crasso
- Mal di pancia e di stomaco: da cosa può dipendere e quali sono le cure
- Mal di pancia forte: quando chiamare il medico?
- Differenza tra ulcera gastrica, duodenale, peptica ed esofagea
- Ulcera peptica: complicanze, cura, dieta, quando è pericolosa
- Ecografia renale: esecuzione, indicazioni, costo
- Ecografia alla vescica vuota o piena: preparazione, costo, svolgimento
- Feci dalla bocca: il vomito fecaloide
- Incontinenza fecale: dieta, Alzheimer, parto, cause e terapie
- Il mio alito odora di feci: cause, quando è pericoloso e rimedi
- Morbo di Crohn: cos’è, cause scatenanti, sintomi, cure e dieta
- Differenze tra ileo meccanico ed ileo paralitico: cause, sintomi e trattamenti
- Colite ulcerosa: cause, diagnosi, cura, dieta, cosa mangiare, rimedi
- Differenza tra colon irritabile, colite e colite spastica: sono la stessa cosa?
Dott. Emilio Alessio Loiacono
Medico Chirurgo
Direttore dello Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o unisciti al nostro gruppo Facebook o ancora seguici su Twitter, su Instagram, su YouTube, su LinkedIn, su Tumblr e su Pinterest, grazie!
