
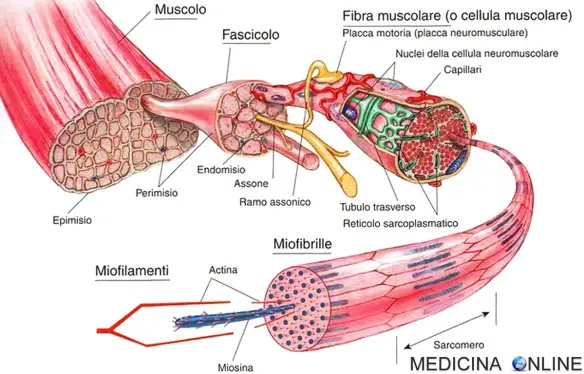
 Con “distrofia muscolare” in medicina si indica un gruppo composto da varie malattie neuromuscolari a carattere degenerativo, caratterizzate da perdita progressiva ed irreversibile di fibra muscolare, determinate geneticamente e che causano atrofia progressiva della muscolatura scheletrica. Pur essendo tra i bambini la distrofia muscolare di Duchenne e quella di Becker le forme più diffuse (specie la Duchenne) e tra gli adulti la distrofia miotonica, esistono in realtà molte altre tipologie intermedie con decorsi estremamente soggettivi e differenti da caso a caso, tra cui:
Con “distrofia muscolare” in medicina si indica un gruppo composto da varie malattie neuromuscolari a carattere degenerativo, caratterizzate da perdita progressiva ed irreversibile di fibra muscolare, determinate geneticamente e che causano atrofia progressiva della muscolatura scheletrica. Pur essendo tra i bambini la distrofia muscolare di Duchenne e quella di Becker le forme più diffuse (specie la Duchenne) e tra gli adulti la distrofia miotonica, esistono in realtà molte altre tipologie intermedie con decorsi estremamente soggettivi e differenti da caso a caso, tra cui:
- Distrofia muscolare di Emery-Dreifuss
- Miopatia X-linked con eccessiva autofagia
- Sindrome di Barth
- Distrofia miotonica di tipo 1 (o di Steinert)
- Distrofia miotonica di tipo 2 (o miopatia miotonica prossimale)
- Distrofia facio-scapolo-omerale
- Distrofia oculofaringea
- Distrofia muscolare scapolo-peroneale
- Miopatia di McLeod
- Distrofia muscolare dei cingoli 1F – LGMD1F
- Distrofia muscolare dei cingoli 2B
- Miopatia nemalinica
- Miopatia di Bethlem
- Sindrome dolorosa regionale complessa
- Distrofie muscolari congenite:
- Distrofia muscolare congenita di Ullrich
- Distrofie da deficit di merosina
- Distrofie da deficit di selenoproteina 1
- Distrofie da alterata glicosilazione dell’alfa-distroglicano (le prime tre sono considerate collegate anche alla distrofia miotonica di tipo 1):
- Distrofia di Fukuyama
- Muscle-Eye-Brain Disease
- Sindrome di Walker-Warburg
- Distrofia MDC1C
- Distrofia MDC1D
- Distrofie distali:
- Miopatia distale di Welander
- Miopatia di Myoshi
- Miopatia distale finlandese tibiale
- Miopatia di Nonaka
- Miopatia di Udd-Markesbery-Griggs
- Miopatia di Gowers-Laing
- Miosite ereditaria da corpi inclusi di tipo 1
- Miopatia distale con indebolimento delle corde vocali e dei muscoli faringei
- Zaspopatia o miopatia distale associata al gene ZASP.
In questo articolo ci occuperemo in particolare delle forme più diffuse tra i bambini (Duchenne e Becker) e tra gli adulti (miotonica).
Distrofia muscolare di Duchenne
Questo tipo di distrofia, detta anche distrofia muscolare generalizzata dell’infanzia, è la più frequente e la meglio conosciuta tra le distrofie muscolari dell’infanzia. Ha un decorso relativamente rapido e attivo. L’incidenza varia da 13 a 33 casi/100.000.
Cause
La patologia è trasmessa come tratto recessivo legato al cromosoma X, si manifesta prevalentemente nei maschi. Nel 30% dei pazienti vi è un’anamnesi familiare negativa e si ritiene che in questi casi avvenga una mutazione spontanea del cromosoma. Si osserva occasionalmente una forma distrofica muscolare prossimale nelle ragazze. Questo fatto può avere molte spiegazioni:
- se la femmina ha un solo cromosoma X;
- per inattivazione del cromosoma X paterno in una grande quantità di cellule embrionali.
L’alterazione del gene localizzato sul cromosoma X determina la mancata produzione di una proteina denominata distrofina. Nel muscolo questa è localizzata sul versante citoplasmatico del sarcolemma dove interagisce con la F-actina del citoscheletro, la struttura filamentosa di rinforzo della cellula muscolare. Inoltre è strettamente legata ad un complesso di proteine sarcolemmali conosciute come proteine legate alla distrofina (DAPs) e glicoproteine legate alla distrofina (DAGs). La mancanza della distrofina conduce ad una perdita delle DAPs e alla rottura del complesso proteina-distroglicano. Questa rottura rende il sarcolemma suscettibile alla lacerazione durante la contrazione muscolare
Cosa accade a livello muscolare?
Negli stadi precoci le caratteristiche principali sono la degenerazione segmentale, la fagocitosi di singole fibre o gruppi di esse, la rigenerazione promossa dalla necrosi. Con il progredire della malattia si osservano modificazioni comuni a tutti i tipi di distrofia muscolare: perdita di fibre muscolari, fibre residue di maggiore o minore diametro rispetto al normale e disposte casualmente, aumento degli adipociti e fibrosi. Si osserva quindi uno stato di ipertrofia, risultato dell’ingrossamento delle fibre sane rispetto alle fibre adiacenti inutilizzate. Successivamente la vera ipertrofia viene sostituita da una pseudoipertrofia, dovuta alla sostituzione delle fibre degenerate con tessuto adiposo. Alla fine le fibre degenerano e scompaiono, presumibilmente a causa dell’estinguersi della capacità di rigenerazione. In quest’ultimo stadio rimangono solo poche fibre muscolari sparse, quasi perse in un mare di adipociti.
Leggi anche:
- Differenza tra distrofia muscolare e sclerosi multipla
- Differenza tra distrofia muscolare e SLA (sclerosi laterale amiotrofica)
- Differenza tra distrofia muscolare ed atrofia muscolare progressiva
- Rabdomiolisi: sintomi, da statine, conseguenze, valori
- CPK in adulti e bambini: alto, basso, conseguenze, cure e rimedi naturali
Sintomi e segni di distrofia di Duchenne
La distrofia di Duchenne viene riconosciuta al terzo anno di vita, quando si nota nel bimbo una incapacità a camminare o correre in modo normale, funzioni che invece – a questa età – dovrebbero già essere ben acquisite. Altri sintomo potrebbe essere il fatto che il bambino, che prima camminava normalmente, negli ultimi tempi tende a cadere più spesso od evita di correre, risultando molto meno attivo rispetto al solito. Con il passare del tempo aumentano nel piccolo paziente le difficoltà a camminare, correre e salire le scale. I primi muscoli ad essere colpiti sono il quadricipite, i glutei. I muscoli della scapola degli arti superiori vengono colpiti successivamente. L’ingrossamento dei polpacci e di altri muscoli è progressivo nei primi stadi della malattia, anche quelli originariamente ingrossati, tendono a ridursi di volume.
Gli arti appaiono gradatamente sempre più ipotonici e flaccidi, e con il progredire della malattia compaiono contratture conseguenti al mantenimento degli arti nella stessa posizione e al mancato bilanciamento fra agonisti ed antagonisti.
I riflessi tendinei diminuiscono fino ad azzerarsi del tutto; gli ultimi a scomparire sono i riflessi achillei. Le ossa divengono sottili e demineralizzate. I muscoli lisci sono risparmiati, mentre il cuore può dar segni di varie tipologie di aritmia. In casi molto rari si osserva un modesto ritardo mentale non progressivo.
Aspettativa di vita e morte
L’aspettativa di vita dipende sempre dal soggetto e negli ultimi quindici anni le prospettive di vita si sono allungate notevolmente grazie alla ventilazione notturna; se nel secolo scorso alcuni medici sostenevano che un paziente affetto da DMD potesse difficilmente superare la seconda decade, oggi ci sono casi di pazienti con Distrofia Muscolare di Duchenne che vivono oltre il cinquantesimo anno di età. Di solito la morte è dovuta ad insufficienza respiratoria, infezioni polmonari o scompenso cardiaco.
Distrofia muscolare di tipo Becker
Altra tipologia di distrofia ben caratterizzata, strettamente correlata alla variante di Duchenne, di cui rappresenta una forma relativamente più benigna. La sua incidenza è difficile da valutare, forse intorno a 3-6 casi/100.000 nati vivi. Si tratta anch’essa di una malattia legata al cromosoma X, trasmessa praticamente solo ai maschi. Al contrario che nella forma di Duchenne, qui la distrofina è presente, ma risulta strutturalmente anormale. Essa provoca debolezza e atrofia degli stessi muscoli coinvolti nella distrofia di Duchenne, ma l’esordio è più tardivo, comparendo intorno ai 12 anni. L’età media alla quale viene persa la capacità di camminare è di 25-30 anni e la morte sopraggiunge in genere nella quinta decade. L’interessamento cardiaco è meno frequente e le facoltà intellettive sono quasi sempre normali.
Distrofia miotonica
E’ la forma di distrofia più diffusa negli adulti e si caratterizza per la trasmissione autosomica dominante ad alta penetranza, per l’associazione con la miotonia e per la presenza di alterazioni distrofiche nei tessuti extramuscolari (cristallino, testicolo, cute, esofago, cuore).
Sintomi
L’atrofia muscolare non si rende evidente fino alla prima età adulta. Sono spesso i piccoli muscoli delle mani a diventare atrofici; in altri casi i segni più precoci sono la ptosi palpebrale e l’assottigliamento e il rilasciamento dei muscoli della faccia. L’atrofia dei masseteri porta ad un assottigliamento della metà inferiore della faccia e alla malposizione della mandibola. Gli sternocleidomastoidei sono quasi sempre assottigliati e indeboliti con conseguente curvatura inferiore del collo (collo a cigno).
La debolezza dei muscoli della faringe e della laringe determina una voce debole e monotona. Frequenti sono la debolezza del diaframma e l’ipoventilazione alveolare con bronchite cronica. Anche le alterazioni cardiache sono frequenti, dovute il più delle volte ad anomalie della conduzione.
Caratteristica peculiare della malattia è la miotonia, che si esprime come contrazione prolungata di certi muscoli dopo una breve percussione o stimolazione elettrica o come ritardo del rilasciamento dopo contrazione volontaria. La miotonia può precedere la debolezza di molti anni.
La malattia progredisce lentamente, con interessamento graduale dei muscoli prossimali degli arti e del tronco. La maggior parte dei pazienti è confinata in carrozzella o a letto entro 15-20 anni dall’esordio e la morte si verifica a causa di infezioni polmonari, blocco cardiaco o scompenso cardio-circolatorio. E un esempio di eterogeneita genetica (è interessato un altro gene responsabile della merosina e non la disrofina come nello DMD e BMD) e hanno lo stesso quadro fenotipico di espressione simili. Sono presenti 4 fenotipi:
- Classico: 60% in paesi occidentali con ipodensità della sostanza bianca e dilatazione dei ventricoli e lo sviluppo intellettivo nella norma.
- Fukuyama: nei paesi asiatici con presenza di attivazione strutturale nel cervello, ritardo normale e epilessia.
- Cerebro-muscolo-oculare: con presenza di disturbi neurologici e oculari e grave ritardo mentale.
- Walker-Warburg: più severa dal p.v. neurologico.
Terapia delle distrofie
Purtroppo, allo stato attuale della ricerca scientifica, non sono note terapie che possano invertire la progressione della patologia, né di bloccarla. La somministrazione di prednisone sembra però quantomeno ritardare la progressione della distrofia fino ad un periodo di tre anni. Le speranze dei ricercatori provengono soprattutto dal tentativo di realizzare una terapia genica, cioè introducendo nell’organismo ammalato copie corrette del gene difettoso. Quattro sono i cardini del trattamento dei pazienti con distrofia muscolare:
- evitare il prolungato riposo a letto (con elevato rischio di lesioni da decubito);
- mobilizzazione passiva con fisioterapista;
- incoraggiare il paziente a condurre il più a lungo una vita normale;
- supporto psicologico al paziente ed alla sua famiglia.
Questo aiuta a prevenire il rapido peggioramento che consegue all’inattività e a mantenere una sana disposizione mentale. Purtroppo in molti casi nelle fasi avanzate della malattia si rende necessario l’uso di supporti per il movimento, come la sedia a ruote, e la respirazione assistita, in quanto l’insufficienza respiratoria è un disturbo subdolo che si può manifestare sotto forma di apnee notturne.
Leggi anche:
- Differenze tra la distrofia muscolare di Duchenne e di Becker
- Distrofia muscolare di Duchenne, di Becker e di Emery-Dreifuss
- Distrofia di Landouzy-Dejerine, oftalmoplegia esterna progressiva, sindrome di Kearns-Sayre e distrofia oculofaringea
- Distrofia miotonica di tipo 1 e 2: cause, trasmissione, sintomi, diagnosi, cure
- Distrofia miotonica di Steinert (tipo 1) e distrofia miotonica congenita
- Miopatia miotonica prossimale (malattia di Ricker): cause, sintomi, diagnosi, cure
- Le malattie genetiche più diffuse al mondo
- Atrofia muscolare progressiva: cause, sintomi, cura, aspettativa di vita
- Differenze tra atrofia muscolare progressiva e sclerosi laterale amiotrofica
- Sclerosi laterale amiotrofica (SLA): cause, sintomi, diagnosi e prognosi
- Differenza tra atrofia, distrofia ed aplasia con esempi
- Sclerosi multipla: cause, sintomi, diagnosi e prognosi
- Differenze tra sclerosi laterale amiotrofica e sclerosi multipla
- Atrofia muscolare spinale: sintomi, trasmissione, tipi e cure
- Sistema nervoso: com’è fatto, a che serve e come funziona
- I muscoli: come sono fatti, come funzionano e cosa rischiano quando ti alleni
- Morbo di Parkinson: cause, sintomi, decorso, terapie
- Morbo di Alzheimer: cause, sintomi, decorso, terapie
- Differenza tra morbo di Alzheimer e morbo di Parkinson: sintomi comuni e diversi
- Differenza tra morbo di Alzheimer, demenza senile, vascolare e reversibile
- Demenza senile: cause, sintomi, decorso e cure
- Differenza tra bicipite, tricipite e quadricipite
- Stiramento muscolare: sintomi, rimedi e prevenzione
- Differenza tra muscoli adduttori e abduttori
- Differenza tra muscoli agonisti, antagonisti e sinergici
- Classificazione dei muscoli
- Differenza tra frattura composta, composta, esposta e patologica
- Callo osseo e pseudoartrosi, quando la frattura non guarisce: cause, diagnosi e terapie
- Femore rotto: tipi di frattura, sintomi, intervento, riabilitazione e conseguenze
- Articolazione del ginocchio: com’è fatta, quali sono le patologie, i sintomi e gli esami da fare ?
- Come riconoscere un atleta “natural” da un dopato in palestra
- Digiuno intermittente e terapeutico: fa dimagrire? Fa bene o fa male alla salute?
- Femore: anatomia e funzioni in sintesi
- Dismetria degli arti inferiori: una gamba è più corta dell’altra
- Cosa sono e qual è la differenza tra massa magra e massa grassa? Tutte le percentuali di grasso, ossa e muscoli
Lo Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o unisciti al nostro gruppo Facebook o ancora seguici su Twitter, su Instagram o su Pinterest, grazie!
