
 La fibrosi cistica polmonare o, più semplicemente, “fibrosi cistica” (acronimo FC, in inglese “cystic fibrosis”) è una rara, debilitante e mortale malattia genetica autosomica recessiva causata da una mutazione di un gene sito sul cromosoma 7, il quale codifica per una proteina che funziona come canale per il cloro, detta CFTR (Cystic Fibrosis Transmembrane conductance Regulator). La fibrosi cistica è la malattia genetica ereditaria mortale più comune. La sintomatologia è complessa, coinvolge numerosi e differenti organi interni ed è tutta riconducibile all’anomalia nell’escrezione del cloro, normalmente mediata dalla proteina che in questa malattia è alterata. L’anomala escrezione del cloro determina la secrezione di muco anomalo, denso, viscoso e poco scorrevole, che tende ad ostruire vari dotti del corpo: tale ostruzione provoca i sintomi e segni principali della malattia, che includono infezioni polmonari ricorrenti e gravi, difficoltà respiratorie, insufficienza del pancreas, pancreatiti, malassorbimento con steatorrea, malnutrizione per difetto, epatopatie come la cirrosi epatica, ostruzione intestinale con stipsi frequente, infertilità maschile, osteoporosi.
La fibrosi cistica polmonare o, più semplicemente, “fibrosi cistica” (acronimo FC, in inglese “cystic fibrosis”) è una rara, debilitante e mortale malattia genetica autosomica recessiva causata da una mutazione di un gene sito sul cromosoma 7, il quale codifica per una proteina che funziona come canale per il cloro, detta CFTR (Cystic Fibrosis Transmembrane conductance Regulator). La fibrosi cistica è la malattia genetica ereditaria mortale più comune. La sintomatologia è complessa, coinvolge numerosi e differenti organi interni ed è tutta riconducibile all’anomalia nell’escrezione del cloro, normalmente mediata dalla proteina che in questa malattia è alterata. L’anomala escrezione del cloro determina la secrezione di muco anomalo, denso, viscoso e poco scorrevole, che tende ad ostruire vari dotti del corpo: tale ostruzione provoca i sintomi e segni principali della malattia, che includono infezioni polmonari ricorrenti e gravi, difficoltà respiratorie, insufficienza del pancreas, pancreatiti, malassorbimento con steatorrea, malnutrizione per difetto, epatopatie come la cirrosi epatica, ostruzione intestinale con stipsi frequente, infertilità maschile, osteoporosi.
La fibrosi cistica, un tempo mortale nel primo anno di nascita, è oggi una malattia che può essere curata, purtuttavia rimane una malattia debilitante, che abbassa la qualità della vita del paziente e che accorcia la vita di quest’ultimo di molti anni.
La diagnosi e la cura della fibrosi cistica coinvolge un gran numero di figure professionali, tra cui: pediatri, genetisti, tecnici di laboratorio, internisti, ortopedici, gastroenterologi, chirurghi generali, endocrinologi, diabetologi, andrologi, medici esperti in riproduzione, psicologi, psichiatri, infermieri, fisioterapisti. La possibilità di condurre una vita comunque dignitosa passa fortemente dalla forza di volontà del paziente e dalla presenza costante dei suoi famigliari e amici.
Epidemiologia
La fibrosi cistica è la più comune malattia autosomica recessiva tra le persone di etnia caucasica. Negli Stati Uniti, circa 30.000 persone ne vivono la condizione. La maggior parte dei casi viene diagnosticata a partire dai sei mesi di età. In Canada, vi sono circa 3.500 persone affette da fibrosi cistica. Circa 1 persona su 25 di discendenza europea e una su 30 degli americani caucasici, è un portatore di una mutazione che causa la fibrosi cistica. Anche se la condizione è meno comune in questi gruppi, circa 1 su 46 ispanici, 1 su 65 africani e 1 su 90 asiatici hanno almeno un gene CFTR anormale. L’Irlanda presenta la più alta incidenza al mondo di fibrosi cistica: 1 su 1353 individui. Anche se è una malattia rara, la fibrosi cistica è classificata come una delle malattie genetiche più frequenti in grado di accorciare la vita e determinare la morte del paziente. È più comune tra le nazioni del mondo occidentale. Un’eccezione è la Finlandia, dove si riscontra solamente una su 80 persone portatori di una mutazione della condizione. Negli Stati Uniti, 1 su 4.000 bambini nascono con la fibrosi cistica. Sempre negli Stati Uniti, nel 1997, circa 1 su 3300 bambini caucasici è nato con la malattia. Al contrario, solo 1 su 15.000 bambini afro-americani soffriva di tale condizione, negli americani asiatici il tasso era ancora più basso: 1 su 32.000. La fibrosi cistica si riscontra nei maschi e nelle femmine allo stesso modo. Per ragioni non ancora chiare, i dati hanno dimostrato che i maschi tendono ad avere una speranza di vita più lunga rispetto alle femmine, tuttavia studi recenti suggeriscono che questo divario non esiste più, probabilmente ciò è dovuto al miglioramento dei servizi sanitari, mentre un recente studio irlandese ha individuato un legame tra l’ormone femminile estrogeno e peggiori condizioni della malattia. La distribuzione degli alleli della fibrosi cistica varia tra le popolazioni. La frequenza di portatori ΔF508 è stata stimata in 1:200 nel nord della Svezia, in 1:143 in Lituania e 1:38 in Danimarca. ΔF508 si verifica in Finlandia, ma è un allele in minoranza. La fibrosi cistica si verifica in solo 20 famiglie finlandesi.
Cause
La fibrosi cistica è causata da una mutazione nel gene CFTR (acronimo di “Cystic Fibrosis Transmembrane Regulator”, cioè “regolatore transmembrana della fibrosi cistica”.
| Mutazione | Frequenza nel mondo |
|---|---|
| ΔF508 | 66%–70% |
| G542X | 2,4% |
| G551D | 1,6% |
| N1303K | 1,3% |
| W1282X | 1,2% |
| Tutti gli altri | 27,5% |
La mutazione più comune, come visibile dallo schema in alto, è quella ΔF508, una delezione di tre nucleotidi che si traduce in una perdita di fenilalanina nella posizione 508a della proteina. Questa mutazione si riscontra nei due terzi (66-70%) di tutti i casi mondiali di fibrosi cistica e il 90% dei casi negli Stati Uniti. Tuttavia, vi sono più di 1500 ulteriori mutazioni che possono condurre alla malattia. Anche se la maggior parte delle persone hanno due copie (alleli) del gene CFTR, una sola è necessaria per evitare la condizione. La fibrosi cistica si sviluppa quando non vi è alcun allele in grado di produrre una proteina CFTR funzionale: la fibrosi cistica è quindi considerata una malattia autosomica recessiva. E’ interessante notare come, in condizione di eterozigosi, la presenza di un solo allele recessivo fornisce al soggetto un vantaggio, rendendolo immune dagli effetti della febbre tifoide.
Gene interessato
Il gene CFTR, si trova al locus q31.2 del cromosoma 7, è composto da 230.000 paia di basi e crea una proteina che è lunga 1.480 aminoacidi. In particolare, la posizione è posta tra le 117.120.016 e le 117.308.718 paia di basi sul braccio lungo del cromosoma 7, regione 3, banda 1, sottobanda 2, rappresentato come 7q31.2. Strutturalmente, il gene CFTR è un tipo di gene noto come gene ABC. Il prodotto di questo gene è un canale di ione cloruro importante nella produzione di sudore, dei succhi digestivi e dell’espettorato.
Trasmissione autosomica recessiva
La fibrosi cistica si trasmette con modalità “autosomica recessiva”. Una malattia è detta a trasmissione autosomica recessiva quando l’allele alterato deve essere presente in coppia (omozigosi), cioè sono necessarie due copie dell’allele difettoso per far sì che la malattia si esprima, a prescindere dal sesso. Non basta un solo genitore portatore sano o malato, bensì entrambi i genitori devono essere portatori sani o malati. Il fenotipo quindi si esprime quando nel genotipo dell’individuo sono presenti entrambi gli alleli responsabili, fatto che spiega l’alta probabilità di sviluppare malattie genetiche in caso di incesto. Quindi:
- un individuo che possegga entrambi gli alleli alterati: è portatore ed è malato;
- un individuo che possegga solo un allele alterato: è portatore ma è sano;
- un individuo che non possegga nessun allele alterato: NON è portatore ed è sano.
Essere portatore sano vuol dire quindi NON avere la patologia ma possedere nel proprio genotipo un allele mutato, che può essere trasmesso alle generazioni successive.
Dalla combinazione delle possibili condizioni di genitori sani, malati e portatori sani, deriva la distribuzione probabilità che la malattia sia trasmessa ai figli:
- genitori malato-malato: la probabilità che il figlio/a nasca malato è del 100%;
- genitori sano-malato: la probabilità che il figlio/a nasca portatore sano è del 100%;
- genitori malato-portatore sano: la probabilità che il figlio/a nasca malato è del 50% e del 50% che nasca portatore sano;
- genitori sano-portatore sano: la probabilità che il figlio/a nasca sano è del 50% e del 50% che nasca portatore sano;
- genitori portatore-portatore: la probabilità che il figlio/a nasca portatore sano è del 50% mentre è del 25% che nasca sano o malato.
Se nessuno dei genitori ha un allele mutato, non c’è ovviamente alcuna trasmissione autosomica recessiva ed i figli saranno tutti sani e NON portatori dell’allele mutato.
Nell’immagine che segue, è raffigurata la tipica situazione in cui entrambi i genitori sono sani ma portatori dell’allele mutato:
- un figlio su quattro avrà entrambi gli alleli alterati e sarà malato ed ovviamente portatore;
- due figli su quattro avranno un allele normale ed uno alterato e saranno sani ma anche portatori;
- un figlio su quattro avrà entrambi gli alleli normali e sarà sano e NON portatore.

Leggi anche:
- Differenza tra malattia autosomica dominante e recessiva con esempi
- Le malattie genetiche più diffuse al mondo
- Sindrome di Turner: cariotipo, cause, sintomi e segni caratteristici
- Sindrome di Klinefelter: cariotipo, cause, sintomi e cura
- Ereditarietà X linked, malattie dominanti e recessive legate al cromosoma X: significato, esempi
- Differenza tra allele dominante e recessivo
- Differenza tra portatore sano e malato in genetica
- Cosa significa “portatore sano” in genetica e nelle infezioni?
- Differenza tra omozigote ed eterozigote
Genotipo e fenotipo
Vi è una certa correlazione tra la mutazione (genotipo) e le manifestazioni cliniche della malattia (fenotipo). La grande variabilità delle manifestazioni e la gravità della malattia sono legate alle molteplici mutazioni nel gene CFTR. La penetranza è di solito del 100% degli omozigoti con mutazioni gravi, ma la gravità della malattia è variabile con forme più lievi, senza insufficienza pancreatica o con moderata insufficienza respiratoria. L’omozigosi ΔF508/ΔF508 è associata con la forma classica della malattia, con un aumento degli elettroliti nel sudore, insufficienza pancreatica e polmonare spesso di forma grave. Alcune mutazioni causano la compromissione di più funzionalità e altri studi epidemiologici mostrano popolazioni differenti con differenti mutazioni, così che le mutazioni ΔF508/R117H, ΔF508/ΔI507, KBC-10 ΔF508/3849 → T e G-5 ΔF508/2789 → A hanno un tasso di mortalità inferiore alla omozigosi F508. Le mutazioni ΔF508/R117H, ΔF508/ΔI507, F508 / 3849 10 KBC → T, G-5 ΔF508/2789 → A ΔF508/A455E hanno manifestazioni fenotipiche minori.
Genotipi esistenti:
| Coppia di mutazioni | Frequenza |
|---|---|
| ΔF508 – ΔF508 (omozigosi) | 50 % |
| ΔF508 – altre mutazioni (eterozigosi) | 40 % |
| altre mutazioni – altre mutazioni | 10 % |
Leggi anche: Differenza tra genotipo e fenotipo
Segni e sintomi
I segni distintivi della fibrosi cistica sono pelle salata, scarsa crescita e scarso aumento di peso nonostante una normale assunzione di cibo, accumulo di muco denso e appiccicoso, infezioni polmonari frequenti e tosse o mancanza di respiro. I maschi possono non essere fertili a causa dell’assenza congenita dei vasi deferenti. I sintomi spesso compaiono durante l’infanzia, come l’ostruzione intestinale a causa di ileo patologico da meconio nei neonati. Quando i bambini crescono, vi sono complicanze nel rilascio del muco negli alveoli. Le cellule ciliate epiteliali del paziente hanno una proteina mutata che porta alla produzione di muco anormalmente viscoso. La scarsa crescita nei bambini si presenta tipicamente come l’incapacità di aumentare di peso o di altezza rispetto ai loro coetanei. Spesso la condizione non viene diagnosticata fino a quando non si cercano le cause di questa scarsa crescita. Le cause della mancata crescita sono multifattoriali e comprendono l’infezione polmonare cronica, il cattivo assorbimento delle sostanze nutrienti attraverso il tratto gastrointestinale e l’aumento della domanda metabolica a causa dello stato cronico di malattia. In rari casi, la fibrosi cistica può manifestarsi come un disturbo della coagulazione del sangue. I bambini piccoli sono particolarmente sensibili ai disturbi da malassorbimento di vitamina K perché solo una piccola quantità di questa vitamina attraversa la placenta, lasciando il bambino con riserve molto basse. Poiché i fattori II, VII, IX e X (fattori di coagulazione) sono vitamina K-dipendenti, bassi livelli di essa possono causare problemi. La patologia polmonare è la conseguenza dell’ostruzione delle vie aeree causata dall’accumulo di muco, dalla riduzione della clearance mucociliare e dall’infiammazione.
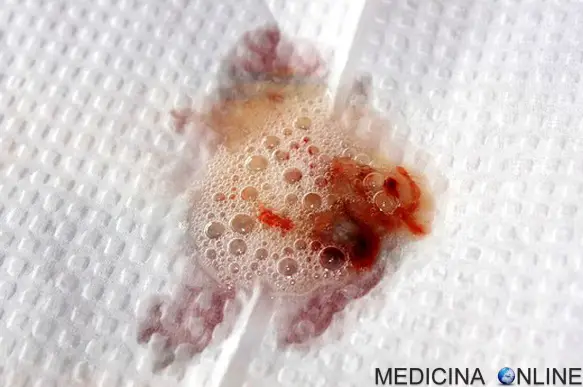
Espettorato con sangue
Polmoni
L’infiammazione e l’infezione causano lesioni e cambiamenti strutturali per i polmoni, portando ad una varietà di sintomi. Nelle fasi iniziali, tosse incessante, produzione abbondante di espettorato e ridotta capacità polmonare, sono condizioni comuni. Molti di questi sintomi si verificano quando i batteri, che normalmente abitano l’espettorato denso, crescono fuori controllo e causano polmonite. Nelle fasi successive, i cambiamenti nella struttura del polmone, come le patologie delle vie aeree principali (bronchiectasie), aggravano ulteriormente le difficoltà nella respirazione. Altri sintomi includono tosse con sangue (emottisi), alta pressione sanguigna nei polmoni (ipertensione polmonare), insufficienza cardiaca, difficoltà ad ottenere abbastanza ossigeno (ipossia) e insufficienza respiratoriache richiede il supporto con maschere respiratorie. Lo Staphylococcus aureus, l’Haemophilus influenzae e il Pseudomonas aeruginosa sono i tre organismi più comuni che causano infezioni polmonari nei pazienti con la fibrosi cistica. Oltre alle tipiche infezioni batteriche, le persone con la condizione, solitamente, sviluppano altri tipi di malattie polmonari. Tra queste l’aspergillosi broncopolmonare allergica, in cui la risposta del corpo al comune fungo Aspergillus fumigatus provoca un peggioramento dei problemi respiratori. Un’altra patologia è l’infezione da Mycobacterium avium complex (MAC), un gruppo di batteri legati alla tubercolosi, che possono causare danni ai polmoni e non rispondono agli antibiotici comuni.
Il muco presente nei seni paranasali è ugualmente denso e può anche causare il blocco dei passaggi, con conseguente infezione. Questo può causare dolore facciale, febbre, scolo nasale, cefalea e aumentare le difficoltà respiratorie. Soggetti con fibrosi cistica possono sviluppare crescita eccessiva del tessuto nasale (Poliposi naso-sinusale) a causa di infiammazione da infezioni croniche dei seni. Polipi ricorrenti possono verificarsi in circa il 10% al 25% dei pazienti affetti da fibrosi cistica. Le complicanze cardiorespiratorie sono la causa più comune di morte (~ 80%) nei pazienti affetti dalla condizione.
Leggi anche:
- Broncopolmonite: sintomi iniziali, contagio, prognosi, morte
- Sclerodermia: cause, sintomi e cura
- Sindrome di Sjögren: sintomi, invalidità, terapia e mortalità
- Fibromialgia: sintomi, cause, cura e tender points
Segni e sintomi gastrointestinali
Prima dello screening prenatale e neonatale, la fibrosi cistica era spesso diagnosticata quando un neonato non era in grado di espellere le feci (meconio). Il meconio può bloccare completamente l’intestino e causare gravi malattie. Questa condizione, detta ileo da meconio, si verifica nel 5-10% dei neonati con fibrosi cistica. Inoltre, la protrusione della membrana interna rettale (prolasso rettale) è più comune, e si verifica in circa il 10% dei bambini affetti dalla condizione, ed è causata da un aumento del volume fecale, dalla malnutrizione e da una maggiore pressione intra-addominale causata dalla tosse.
Pancreas
Le secrezioni anomale nel pancreas bloccano il movimento degli enzimi digestivi nel duodeno e provocano danni irreversibili al pancreas, spesso sfociando in una dolorosa infiammazione (pancreatite). Nei casi più gravi e avanzati, i dotti pancreatici appaiono atrofici.
Leggi anche:
- Pancreatite acuta: terapia, dieta, complicanze, morte
- Pancreatite cronica: sintomi, diagnosi, cura, dieta, mortalità
L’insufficienza pancreatica esocrina si verifica nella maggior parte (dall’85% al 90%) dei pazienti con fibrosi cistica. È principalmente associata con “gravi” mutazioni del gene CFTR, in cui entrambi gli alleli sono completamente non funzionali (ad es ΔF508/ΔF508). Si verifica nel 10-15% dei pazienti con una mutazione “grave” e una “media” del gene CFTR, dove c’è ancora una lieve attività dell’CFTR o dove vi sono due “medie” mutazioni. In questi casi più lievi, vi è ancora sufficiente funzione esocrina del pancreas in modo che la supplementazione di enzimi non sia necessaria.
Leggi anche: Insufficienza pancreatica: esami, cause, sintomi, cura, dieta
Le secrezioni dense inoltre possono causare problemi al fegato. La bile secreta per aiutare la digestione può bloccare i dotti biliari, causando danni epatici. Nel corso del tempo, questo può portare a cicatrici e nodularità (cirrosi). Il fegato non riesce a liberare il sangue dalle tossine e non sintetizza le proteine importanti, come quelle responsabili della coagulazione del sangue. Le patologie epatiche sono la terza causa più comune di morte correlata alla fibrosi cistica.
Leggi anche: Cirrosi epatica e fegato: sintomi, dieta, diagnosi, terapia e prevenzione
Oltre ai problemi del pancreas, le persone con fibrosi cistica lamentano bruciore di stomaco, blocco intestinale da intussuscezione e costipazione. Gli individui anziani affetti da fibrosi cistica possono sviluppare ostruzione intestinale distale a causa delle feci ispessite.
Leggi anche:
- Differenze tra ileo meccanico ed ileo paralitico: cause, sintomi e trattamenti
- Fecaloma: tappo di feci durissime, cause, sintomi e rimedi
- Defecografia: cos’è, a che serve, come ci si prepara, è dolorosa?
- Fecaloma ed ostruzione intestinale: quando chiamare il medico
- Feci dure, stitichezza e dolore defecazione: cause e cure
Malnutrizione
La mancanza di enzimi digestivi porta a difficoltà di assorbire i nutrienti, con la loro successiva escrezione nelle feci: il “malassorbimento”. Il malassorbimento porta alla malnutrizione per difetto e alla scarsa crescita. L’ipoproteinemia risultante può essere abbastanza grave da provocare edema generalizzato. Gli individui affetti da fibrosi cistica hanno anche difficoltà di assorbire le vitamine liposolubili A, D, E e K.
Leggi anche:
- Malnutrizione per difetto o eccesso: definizione, sintomi, significato
- Ipovitaminosi (carenza di vitamine): sintomi, lingua, unghie, cura
- Carenza di vitamina A (retinolo): sintomi, dieta e integratori consigliati
- Carenza di vitamina D, rachitismo, osteomalacia: cause, sintomi, terapie, dieta, integratori
- Carenza di vitamina K: sintomi, fabbisogno, dieta e integratori consigliati
- Carenza di niacina e triptofano: pellagra, cause, terapie e dieta
- Carenza e dipendenza da vitamina B6: cause, sintomi, terapie, dieta, integratori
- Carenza di biotina: cause, sintomi, fabbisogno, dieta, integratori
- Carenza di vitamina C e scorbuto: cause, sintomi, cura, dieta, integratori
- Carenza di acido pantotenico (vitamina B5): cause, sintomi, cure, dieta, integratore
- Ipoproteinemia (basso livello di proteine nel sangue): cause e conseguenze
- Iperproteinemia (alto livello di proteine nel sangue): cause e conseguenze
Diabete nella fibrosi cistica
I danni del pancreas possono portare alla perdita delle cellule insulari, causando una forma di diabete caratteristica degli affetti di fibrosi cistica. Ciò è una delle più importanti complicanze non polmonari della malattia. Il diabete è la complicanza non-polmonare più frequente nei casi di fibrosi cistica. Viene riconosciuto come una entità distinta, e presenta caratteristiche miste del tipo 1 e del tipo 2. Nonostante sia utilizzati farmaci orali contro il diabete, l’unico trattamento raccomandato consiste in iniezioni di insulina o l’uso di una pompa di insulina. A differenza del diabete classico, non vengono raccomandate restrizioni nella dieta.
Leggi anche:
- Differenze tra il diabete di tipo 1 e 2 (insulino dipendente e resistente)
- Diabete mellito: diffusione, sintomi, classificazione e diagnosi differenziale
- Diabete mellito: cause e fattori di rischio del tipo 1 e 2
- Diabete mellito: come si forma la malattia?
- Diabete mellito: conseguenze e complicanze a lungo termine
- Prevenzione del diabete mellito
Vitamina D, osteoporosi e dita ippocratiche
La vitamina D è coinvolta nella regolazione del calcio e del fosfato. Lo scarso assorbimento della vitamina D nella dieta, a causa del malassorbimento, può portare a osteoporosi, una condizione in cui le ossa indebolite sono più soggette a fratture. Inoltre, le persone con fibrosi cistica sviluppano spesso le cosiddette dita ippocratiche (dette anche clubbing) a causa del basso tenore di ossigeno nei loro tessuti; a tal proposito, leggi anche: Dita ippocratiche congenite e secondarie: cause, sintomi e terapie

Dita ippocratiche
Infertilità
L’infertilità interessa sia gli uomini che le donne. Almeno il 97% degli uomini affetti da fibrosi cistica sono infertili, ma non sterili e possono avere figli con tecniche di procreazione assistita. La principale causa di infertilità negli uomini affetti da fibrosi cistica è l’assenza congenita dei vasi deferenti (che normalmente connettono i testicoli ai dotti eiaculatori del pene), ma potenzialmente vi possono essere anche ulteriori problemi che possono causare azoospermia, teratospermia e oligoastenospermia. Alcune donne hanno difficoltà di procreazione a causa dell’ispessimento del muco cervicale o per via della malnutrizione. Nei casi più gravi, la malnutrizione interrompe l’ovulazione e causa amenorrea. Leggi anche:
- Differenza tra infertilità e sterilità
- Astenospermia: spermiogramma, spermatozoi deboli e fattori che influenzano la loro motilità
- Ipoposia: quando lo sperma è troppo poco. Cause e terapie per aumentare la quantità di eiaculato
Diagnosi
La fibrosi cistica può essere diagnosticata grazie a molti metodi diversi, tra cui lo screening neonatale, il test del sudore e test genetici. A partire dal 2006 negli Stati Uniti, il 10% dei casi vengono diagnosticati poco dopo la nascita nel quadro di programmi di screening neonatale. Il test inizia con la valutazione della concentrazione sanguigna del tripsinogeno immunoreattivo. Ai neonati con un dosaggio anormale viene effettuato un test del sudore per confermare la diagnosi di fibrosi cistica. I livelli di tripsinogeno possono risultare aumentati negli individui che hanno una sola copia mutata del gene CFTR o, in rari casi, in individui con due copie normali del gene CFTR. A causa di questi falsi positivi, l’utilità dello screening della fibrosi cistica nei neonati è controversa. La maggior parte degli stati e paesi non effettua screening di routine al momento della nascita, di conseguenza, la maggior parte delle persone ricevono una diagnosi dopo che si sono manifestati i sintomi. La forma più comunemente usata per la diagnosi è il test del sudore. Esso comporta l’applicazione di un farmaco che stimola la sudorazione (pilocarpina). Il sudore risultante viene poi raccolto su carta o in un tubo capillare e analizzato per valori anomali di sodio e cloro. Gli individui affetti da fibrosi cistica hanno alti valori di questi due elementi nel sudore. Al contrario, le persone con fibrosi cistica hanno meno tiocianato e isotiocianato nella loro saliva. La fibrosi cistica può essere anche diagnosticata attraverso l’identificazione di mutazioni nel gene CFTR. Le persone con la malattia solitamente sono inserite in un registro che permette ai ricercatori e ai medici di monitorare i risultati di salute e individuare i candidati per le sperimentazioni cliniche.
Diagnosi prenatale
Le coppie che aspettano un bambino o che stanno pianificando una gravidanza, possono farsi testare per le mutazioni del gene CFTR per determinare il rischio che il loro bambino possa nascere affetto da fibrosi cistica. Il test viene tipicamente eseguito prima su uno o su entrambi i genitori e, se il rischio è alto, vengono effettuate prove sul feto. L’American College of Obstetricians and Gynecologists raccomanda il test per le coppie che hanno una storia personale o familiare della malattia. Poiché lo sviluppo della fibrosi cistica nel feto richiede che ogni genitore trasmetta una copia mutata del gene CFTR e poiché il test è costoso, esso viene spesso eseguito inizialmente su di un unico genitore. Se il test rivela che uno dei genitori è un portatore del gene CFTR mutato, l’altro genitore viene testato per calcolare il rischio che i loro figli avranno la malattia. La fibrosi cistica può derivare da più di mille mutazioni differenti. Il test generico analizza il sangue per le mutazioni più comuni, la maggior parte di essi disponibili in commercio cercano 32 o meno differenti mutazioni. Se una famiglia ha una nota rara mutazione, uno screening specifico per tale mutazione può essere eseguito. Poiché non tutte le mutazioni note si trovano sui test attuali, una valutazione negativa del rischio non garantisce che un bambino non avrà la fibrosi cistica. Durante la gravidanza, il test può essere eseguito sulla placenta (villi coriali) o sul liquido intorno al feto (amniocentesi). Tuttavia, il prelievo dei villi coriali ha un rischio di morte fetale di 1 su 100, mentre l’amniocentesi è di 1 su 200. Uno studio recente ha indicato che questo può essere molto più basso, circa 1 su 1600.
Leggi anche:
- Talassemia: cos’è, sintomi, cure, differenti tipi ed alimentazione
- Celiachia: cos’è il glutine, in quali alimenti è contenuto ed in quali no?
- Sindrome di Noonan: cause, sintomi nel neonato, aspettative di vita
- Sindrome di Bloom: cause, sintomi, diagnosi e terapia
Prognosi ed aspettativa di vita
La prognosi per la fibrosi cistica è migliorata grazie alla diagnosi precoce attraverso lo screening, un migliore trattamento e grazie all’accesso alle cure sanitarie. Nel 1959, l’età media di sopravvivenza dei bambini affetti da fibrosi cistica negli Stati Uniti era di sei mesi. Nel 2008, la sopravvivenza media è di 37,4 anni. In Canada, la sopravvivenza media è aumentata dai 24 anni del 1982 a 47,7 anni nel 2007. Degli individui con fibrosi cistica che hanno più di 18 anni a partire dal 2009, il 92% possedeva il diploma di scuola superiore, il 67% aveva avuto almeno qualche istruzione universitaria, il 15% era considerato disabile e il 9% era disoccupato. Il 56% era single mentre il 39% era sposati o viveva con un partner. In Russial’età globale mediana dei pazienti è di 25 anni, ciò a causa dalla mancanza o del costo elevato dei farmaci e perché il trapianto di polmone non viene eseguito.
Trattamento
Nonostante non vi sia alcuna cura per la fibrosi cistica esistono diversi metodi di trattamento. La gestione della malattia è migliorata in modo significativo nel corso degli ultimi 70 anni. Per i bambini nati oltre 70 anni fa con la condizione sarebbe stato improbabile poter vivere oltre il primo anno, mentre oggigiorno hanno una buona probabilità di raggiungere l’età adulta. I recenti progressi nelle tecniche di trattamento hanno fatto sì che un individuo con la malattia possa trascorrere un’esistenza sempre meno gravata dalla patologia. I capisaldi della gestione sono il trattamento delle infezioni delle vie aeree, l’incoraggiamento in una buona alimentazione e uno stile di vita attivo, oltre ad alcune norme comportamentali, come l’evitamento della vicinanza fisica con altri soggetti con fibrosi cistica ed infezioni polmonari.
La gestione della condizione deve essere protratta per tutta la vita del paziente, ed è volta a massimizzare la funzionalità degli organi e di conseguenza la qualità della vita. Gli obiettivi principali della terapia sono i polmoni, il tratto gastrointestinale (comprensivo della terapia supplementare per gli enzimi pancreatici), gli organi riproduttivi (tra cui tecniche di riproduzione assistita) e il supporto psicologico.
L’aspetto più consistente della terapia della fibrosi cistica è la limitazione e il trattamento del danno polmonare causato dall’espettorato denso e dalle conseguenti infezioni. Antibiotici sono prescritti per trattare le infezioni croniche e acute. Dispositivi meccanici di inalazione e farmaci vengono utilizzati per eliminare il muco ispessito. Queste terapie, pur essendo efficaci, possono risultare particolarmente lunghe per il paziente. Una delle difficoltà più importanti che il paziente deve affrontare è quella di trovare il tempo per seguire le cure prescritte. Inoltre, terapie come il trapianto e la terapia genica mirano a curare alcuni degli effetti della fibrosi cistica. La terapia genica punta a introdurre il gene CFTR normale delle vie aeree. Vi sono due meccanismi utilizzabili per l’introduzione del gene CFTR: il primo utilizza un vettore virale (l’adenovirus) e il secondo ricorre all’uso dei liposomi.
Leggi anche:
- Artrite reumatoide: sintomi iniziali, cause, cure e mortalità
- Differenze tra artrite ed artrosi: sintomi comuni e diversi
- Lupus eritematoso sistemico (LES): cause, sintomi e terapie
Antibiotici
Molti pazienti affetti da fibrosi cistica devono assumere antibiotici costantemente allo scopo di prevenire le infezioni. Gli antibiotici sono assolutamente necessari ogni volta che si sospetta una polmonite o si nota un notevole declino della funzionalità polmonare. Una terapia prolungata può rendere necessario un ricovero in ospedale e l’inserimento di un catetere centrale permanente. La terapia inalatoria con antibiotici come tobramicina, colistina e aztreonam viene spesso prescritta per mesi al fine di migliorare la funzionalità polmonare impedendo la crescita di batteri. Gli antibiotici orali, come la ciprofloxacina o l’azitromicina vengono somministrati per aiutare a prevenire l’infezione o per controllare l’eventuale infezione in corso. Gli antibiotici aminoglicosidici (ad esempio la tobramicina) possono causare la perdita dell’udito, danni al sistema dell’equilibrio nell’orecchio interno o problemi renali a lungo termine. Per evitare il verificarsi di questi effetti collaterali, la quantità di antibiotici nel sangue deve essere misurata di routine e regolata di conseguenza.
Trapianto di polmone
Il trapianto di polmone può rendersi necessario negli individui affetti da fibrosi cistica, diminuendo la funzionalità polmonare diminuisce la tolleranza allo sforzo. Sebbene il trapianto singolo di polmone è possibile per altre condizioni, gli individui affetti da fibrosi cistica devono sostituire entrambi i polmoni poiché il polmone rimanente può contenere batteri che possono infettare il polmone trapiantato. Un trapianto di pancreas o di fegato può essere eseguita nello stesso momento, al fine di alleviare i deficit epatici e/o il diabete. Il trapianto polmonare viene consigliato quando la funzionalità polmonare declina al punto in cui è viene richiesta l’assistenza di dispositivi meccanici o la sopravvivenza del paziente viene minacciata.
Leggi anche:
- Sindrome di Down: cause, sintomi in gravidanza e nei neonati
- Malattia di Huntington: cos’è, ereditarietà, come si trasmette, età di insorgenza
- Anemia falciforme: cosa significa, cause, sintomi e cure
- Differenze tra la distrofia muscolare di Duchenne e di Becker
Altri aspetti
I neonati con ostruzione intestinale richiedono un intervento chirurgico, mentre agli adulti con sindrome da ostruzione intestinale distale non viene proposto. Il trattamento dell’insufficienza pancreatica, mediante sostituzione dei mancanti enzimi digestivi nel duodeno, consente di assorbire correttamente le sostanze nutritive e le vitamine che altrimenti andrebbero perse nelle feci.
Lo sviluppo di osteoporosi può essere prevenuto con l’aumento dell’assunzione di vitamina D e di calcio. I pazienti possono essere trattati anche con bifosfonati, anche se gli effetti negativi possono rappresentare un problema. Le difficoltà nella crescita possono essere evitate grazie all’inserimento di un sondino per l’alimentazione al fine di aumentare il numero di calorie introdotte, oppure si può ricorrere alla somministrazione dell’ormone della crescita.
Infezioni dei seni paranasali sono trattate con cicli prolungati di antibiotici. Lo sviluppo di polipi nasali può seriamente limitare il flusso d’aria attraverso il naso, e col tempo ridurre il senso dell’odore. La chirurgia dei seni è spesso utilizzata per alleviare l’ostruzione nasale e per limitare ulteriori infezioni. Per diminuire l’infiammazione nasale possono essere prescritti steroidi come il fluticasone.
L’infertilità nella donna può essere superata grazie alle tecniche di riproduzione assistita, in particolare grazie al trasferimento embrionale. L’infertilità maschile causata dall’assenza dei vasi deferenti può essere ovviata con l’estrazione testicolare dello sperma (TEST) Se il campione raccolto contiene un numero troppo ridotto di spermatozooi per avere sufficienti chance di una fecondazione spontanea, si può ricorrere all’iniezione intracitoplasmatica dello spermatozoo.
Curiosità
Il tema della fibrosi cistica è stato recentemente trattato nel film del 2019 “A un metro da te” (Five Feet Apart), diretto da Justin Baldoni con Haley Lu Richardson e Cole Sprouse nella parte di due giovani malati appunto di fibrosi cistica che si innamorano uno dell’altra. Il film, per la verità, presenza numerose “licenze creative” che sono dei veri e propri errori dal punto di vista medico, come ad esempio l’eccessivo contatto tra i due protagonisti, impossibile e pericolosissimo nella realtà nelle loro condizioni.
Per aprrofondire:
- Fibrosi cistica: storia, organi coinvolti, cause, trasmissione, anatomia patologica
- Fibrosi cistica: sintomi, segni, diagnosi, esami
- Fibrosi cistica: terapia farmacologica e chirurgica, prognosi, mortalità
Leggi anche:
- Cos’è un cromosoma ed a che serve?
- Differenza tra allele dominante e recessivo
- Differenza tra omozigote ed eterozigote
- Differenza tra gene e allele
- Differenza tra genotipo e fenotipo
- Quanti cromosomi hanno esseri umani, scimmie, cani, gatti e topi?
- Quanti cromosomi ha chi è affetto da Sindrome di Down?
- Cos’è un gene ed a che serve?
- Cosa sono gli alleli ed a che servono?
- Differenza tra cellule eucariote e procariote
- Virus e virioni: cosa sono, come sono fatti, come funzionano e come si riproducono
- Differenza tra cellula aploide e diploide con esempi
- Riproduzione cellulare e ciclo cellulare
- Meiosi: spiegazione di tutte tappe
- Mitosi: spiegazione delle quattro fasi
- Differenza tra mitocondri e cloroplasti
- Differenza tra citosol e citoplasma
- Differenza tra virus e batteri: chi è più pericoloso? Diagnosi, sintomi e terapia
- Organelli (organuli) citoplasmatici della cellula animale: cosa sono ed a che servono?
- Mitocondri: definizione, dimensioni e funzioni
- Citoscheletro: funzioni e struttura
- Ribosomi e reticolo endoplasmatico: cosa sono e che funzioni svolgono?
- Nucleo cellulare: funzioni, dimensioni e membrane nucleari
- Lisosomi: cosa sono? Significato e dimensioni
- Perossisomi: definizione e funzioni
- Membrana plasmatica: definizione e funzioni
- Apparato del Golgi: spiegazione semplice e funzioni
- Citosol: definizione e funzioni
Dott. Emilio Alessio Loiacono
Medico Chirurgo
Direttore dello Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o unisciti al nostro gruppo Facebook o ancora seguici su Twitter, su Instagram, su Mastodon, su Tumblr e su Pinterest, grazie!
