
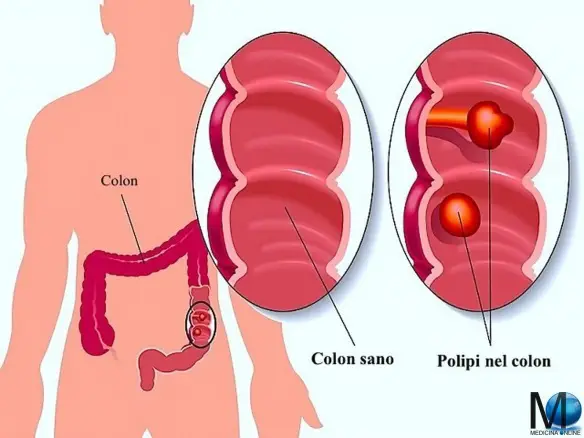
 Con malattia di Cronkhite-Canada (anche chiamata “sindrome di Cronkhite-Canada” o “malattia di Cronkhite” o “sindrome di Cronkhite”; in inglese “Cronkhite – Canada syndrome” o “gastrointestinal polyposis-skin pigmentation-alopecia-fingernail changes syndrome“), in medicina si intende una rara forma di poliposi giovanile a livello gastrointestinale caratterizzata da numerosi polipi a vari livelli del tubo digerente, accompagnata da altre deformazioni. Un polipo gastrointestinale è una crescita anormale di tessuto che sporge dalla mucosa gastroenterica verso il lume di un organo (ad esempio stomaco o intestino). I polipi mostrano una natura amartomatosa. La malattia di Cronkhite-Canada è “sporadica” (cioè non sembra essere una malattia ereditaria) ed è attualmente considerata acquisita e non congenita (cioè non è già presente alla nascita ma si presenta successivamente).
Con malattia di Cronkhite-Canada (anche chiamata “sindrome di Cronkhite-Canada” o “malattia di Cronkhite” o “sindrome di Cronkhite”; in inglese “Cronkhite – Canada syndrome” o “gastrointestinal polyposis-skin pigmentation-alopecia-fingernail changes syndrome“), in medicina si intende una rara forma di poliposi giovanile a livello gastrointestinale caratterizzata da numerosi polipi a vari livelli del tubo digerente, accompagnata da altre deformazioni. Un polipo gastrointestinale è una crescita anormale di tessuto che sporge dalla mucosa gastroenterica verso il lume di un organo (ad esempio stomaco o intestino). I polipi mostrano una natura amartomatosa. La malattia di Cronkhite-Canada è “sporadica” (cioè non sembra essere una malattia ereditaria) ed è attualmente considerata acquisita e non congenita (cioè non è già presente alla nascita ma si presenta successivamente).
Cenni storici
La malattia deve il suo nome al medico di medicina interna Leonard Wolsey Cronkhite Jr. ed alla radiologa Wilma Jeanne Canada, che la descrissero nel 1955.
Epidemiologia
La malattia di Cronkhite-Canada è molto rara. Circa due terzi dei pazienti è di origine giapponese ed il rapporto tra maschio e femmina è 2:1.
Tipologia dei polipi
I polipi si trovano in tutto il tratto gastrointestinale (più frequentemente nello stomaco e nell’intestino crasso, seguiti dall’intestino tenue) ed in genere non si ritrovano nell’esofago. Una biopsia rivela che sono amartomi. La possibilità che progrediscano verso il cancro è generalmente considerata bassa, sebbene sia stata segnalata più volte in passato: i polipi della malattia di Cronkhite-Canada sono quindi considerabili lesioni precancerose.
Cause e fattori di rischio
Le cause della malattia di Cronkhite-Canada sono attualmente ignote: per tale motivo la malattia di Cronkhite-Canada è considerata una malattia “idiopatica“. Inizialmente si era pensato che i cambiamenti epidermici fossero secondari a una profonda malnutrizione a causa dell’enteropatia con perdita di proteine. Recenti scoperte hanno messo in discussione questa ipotesi; in particolare, i cambiamenti di capelli e unghie potrebbero non migliorare con una migliore alimentazione.
Sintomi e segni
I sintomi e segni clinici della malattia includono anomalie variabili dei tessuti ectodermici, tra cui:
- alopecia (caduta dei capelli);
- assottigliamento della cute;
- pigmentazione della pelle;
- atrofia soprattutto a livello delle unghie.
Si osservano spesso inoltre diarrea cronica ed enteropatia con perdita di proteine.
Diagnosi
Non esiste un test specifico per diagnosticare la sindrome di Cronkhite-Canada. La diagnosi si basa su anamnesi, esame obiettivo e per esclusione tramite endoscopia. Gli strumenti che possono essere usati per la diagnosi (e per la diagnosi differenziale), sono:
- anamnesi (raccolta dei sintomi del paziente e della sua storia clinica);
- esame obiettivo (raccolta dei segni, in particolare esame obiettivo addominale e toracico);
- esame del sangue venoso;
- indagini di laboratorio per la ricerca di Citomegalovirus e Helicobacter Pylori;
- esami del sangue per i marker tumorali come l’antigene carcino-embrionale (CEA) e l’antigene carboidratico (CA 19-9);
- radiografia del torace con mezzo di contrasto (pasto di bario);
- radiografia addominale;
- ecografia addominale;
- breath test;
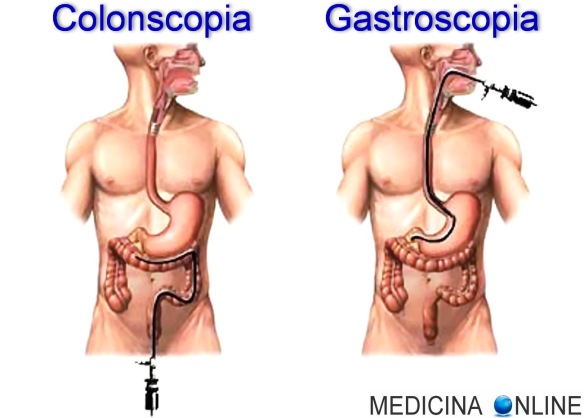
- esame endoscopico (esofagogastroduodenoscopia e/o colonscopia);
- biopsia durante l’esame endoscopico con analisi istologica del campione;
- TC (tomografia computerizzata);
- PET (tomografia ad emissione di positroni);
- ecografia endoscopica (ecoendoscopia).
Non tutti gli esami elencati sono sempre necessari. In genere anamnesi, esame obiettivo, radiologico ed endoscopico con biopsia sono sufficienti per raggiungere una diagnosi.
Diagnosi differenziale
La diagnosi differenziale si pone nei confronti di altre condizioni costituite da più polipi amartomatosi del tratto digestivo, che includono: la sindrome di Peutz – Jeghers, la poliposi giovanile e la malattia di Cowden. Le condizioni di poliposi correlate sono la poliposi adenomatosa familiare, la poliposi adenomatosa familiare attenuata, la sindrome di Birt – Hogg – Dubé e MUTYH.
Terapia
Il supporto nutrizionale è fondamentale e può includere indicazioni dietetiche, integratori, alimentazione in provetta o soluzioni endovenose. I trattamenti proposti comprendono il sodio cromoglicato e l’antinfiammatorio steroideo prednisone, nonché gli antagonisti del recettore dell’istamina (H2) e/o gli inibitori della pompa protonica. Il sodio cromoglicato è in grado di stabilizzare i mastociti determinando un miglioramento in alcune patologie, in particolare di natura allergica.
Leggi anche:
- Sindrome di Zollinger-Ellison (gastrinoma): cause, sintomi, terapia, sopravvivenza
- Gastrite ipertrofica gigante (malattia di Ménétrier): cause, sintomi, diagnosi, cure
- Gastrite acuta e cronica: cause, fattori di rischio, sintomi, segni, complicanze
- Gastrite acuta e cronica: esami, diagnosi, terapia, consigli, dieta, cibi da evitare
- Gastrite cronica, quando il bruciore di stomaco non dà tregua
- Differenza tra polipo e tumore
- Polipi intestinali e polipectomia: come si esegue, biopsia e pericoli
- Differenza tra colonscopia e gastroscopia
- Tumore dello stomaco: sintomi iniziali, sopravvivenza, cure
- Acidità di stomaco e bruciore: tutti i farmaci antiacidi
- Bruciore di stomaco: cosa mangiare, come dormire e rimedi naturali
- Acidità di stomaco: come combatterla con i farmaci antiacidi
- Gastrina alta, bassa, valori, funzioni, cos’è e dove è prodotta
- Bruciore retrosternale (pirosi): cause, sintomi, diagnosi, terapie, rischi
- Inibitori della pompa protonica: effetti collaterali e meccanismo d’azione
- I migliori farmaci antiacidi da banco, senza ricetta medica
- Farmaci procinetici: effetti collaterali e meccanismo d’azione
- Si può vivere senza stomaco? Conseguenze della gastrectomia
- Reflusso gastroesofageo: sintomi, diagnosi e cura
- Reflusso gastroesofageo: dieta, stress, alcolici, latte e notte
- Reflusso gastroesofageo: terapia farmacologica e chirurgica
- Esofago di Barrett, tumore e reflusso gastroesofageo
- Esofago di Barrett: sintomi iniziali, diagnosi, terapia, dieta e chirurgia
- Differenza tra metaplasia, displasia e neoplasia con esempi
- L’apparato digerente: cos’è, com’è fatto, a che serve e come funziona?
- Esofago: anatomia e funzioni in sintesi
- Stomaco: anatomia e funzioni in sintesi
- Capacità massima dello stomaco: si può “mangiare fino a scoppiare”?
- Disfagia nell’anziano: definizione, epidemiologia, cause e fattori di rischio
- Disfagia nell’anziano: sintomi, segni, diagnosi, esami, trattamento
- Differenza tra disfagia di tipo ostruttivo e di tipo motorio
- Differenza tra disfagia ai liquidi e ai solidi
- Differenza tra disfagia ed odinofagia: cause comuni e diverse
- Differenza tra disfagia orofaringea ed esofagea: sintomi comuni e diversi
- Differenza tra disfagia ostruttiva ed occlusione intestinale
- Alimentazione e disfagia nel paziente con morbo di Parkinson
- Polmonite ab ingestis: cause, tempi di guarigione, morte, sopravvivenza
- Inalazione di cibo e corpi estranei nelle vie aeree: sintomi e cosa fare
- Asfissia: sintomi, cure ed in quanto tempo si muore
- Soffocamento: definizione, cause, sintomi, morte
- Morte per soffocamento: segni, sintomi, fasi e tempi
- Soffocamento da cibo, liquidi, saliva in bimbi e adulti: cosa fare?
- Acalasia esofagea: cause, sintomi, cure e prevenzione
- Ritirato in tutta Italia il famoso farmaco anti reflusso: ecco i lotti interessati
- Manometria esofagea: preparazione, esecuzione, rischi, prezzo
- Manometria esofagea 24H: cos’è, a che serve, quando è necessaria
- Manometria esofagea ad alta risoluzione (HRM): cos’è, a che serve, quando è necessaria
- pH-metria esofagea e gastrica: preparazione, esecuzione, rischi, prezzo
- Laringoscopia diretta e indiretta: anestesia, costo, è dolorosa?
- L’esofagogastroduodenoscopia: cos’è, preparazione, è dolorosa o pericolosa?
- Colangiopancreatografia retrograda (ERCP): cos’è, preparazione, è dolorosa o pericolosa?
- Differenza tra ulcera gastrica, duodenale, peptica ed esofagea
- Ulcera peptica: complicanze, cura, dieta, quando è pericolosa
- Cosa succede al cibo nello stomaco dopo averlo ingerito?
- Peristalsi intestinale ed antiperistalsi: caratteristiche e funzioni
- Fundoplicatio secondo Nissen-Rossetti: intervento e rischi
- Acido cloridrico e succo gastrico dello stomaco: di cosa è fatto ed a che serve
- Meccanismi e controllo della secrezione acida dello stomaco
- Dispepsia: cos’è, sintomi, come si fa la diagnosi e terapia
- Stomaco: come fa a digerire il cibo che mangi ed a dirti che sei “pieno”
- Come vincere l’ansia per evitare di mangiare fuori pasto
- Eliminare la tensione nervosa allo stomaco con i rimedi naturali
- Incontinenza fecale primitiva e secondaria: cos’è e come si cura
- Vomitare sangue ed ematemesi: cos’è, cosa fare, cause e terapie
- Da cosa viene causata l’ulcera allo stomaco?
- Feci nere e melena: cause e cure in adulti e neonati
- Breath test Helicobacter: come funziona, come si fa e valori
- Infezione da Helicobacter Pylori: cosa causa, come si riconosce e cura
- Differenza tra tumore benigno, maligno, neoplasia, cancro e metastasi
- Cos’è un tumore? Perché viene il cancro? Quali sono le cause?
- Come nasce un cancro? Cosa sono i cancerogeni e come avviene la cancerogenesi?
- Come prevenire i tumori ed il cancro? I 10 cambiamenti consigliati
- Differenza tra tumore e tessuto normale con esempi
- Cosa sono le metastasi? Tutti i tumori danno metastasi?
- Che significa malattia terminale?
- Stadiazione e classificazione TNM: cancro curabile o terminale?
- Cancro dell’esofago: epidemiologia, età di esordio, tipologie
- Cancro dell’esofago: cause, fattori di rischio, esofago di Barrett
- Cancro dell’esofago: sintomi e segni iniziali, tardivi e di metastasi
- Cancro dell’esofago: esami, diagnosi, stadiazione, classificazione TNM, gravità
- Cancro dell’esofago: trattamento, chirurgia, chemioterapia, radioterapia
- Cancro dell’esofago: prognosi, mortalità, sopravvivenza, aspettativa di vita
- Cancro dell’esofago: prevenzione, dieta, cibi da evitare, consigli
- Cancro dello stomaco: epidemiologia, età di esordio, tipologie
- Cancro dello stomaco: cause, fattori di rischio, fattori protettivi
- Cancro dello stomaco: sintomi e segni iniziali, tardivi e di metastasi
- Cancro dello stomaco: esami, diagnosi, stadiazione, classificazione TNM, gravità
- Cancro dello stomaco: trattamento, chirurgia, chemioterapia, radioterapia
- Cancro dello stomaco: prognosi, mortalità, sopravvivenza, aspettativa di vita
- Cancro dello stomaco: prevenzione, dieta, cibi da evitare, consigli
- Carcinoma precoce dello stomaco (early gastric cancer): cause, sintomi, diagnosi, cure
- Tumore del colon retto: sintomi iniziali, tardivi e ritardo nella diagnosi
- Tumore del colon retto: diagnosi, metastasi, prognosi e stadiazione
- Tumore del colon retto: trattamento chirurgico, radioterapia e chemioterapia
- Tumore del colon retto con metastasi: chirurgia, chemioterapia e terapie biologiche
- Tumore del colon retto: terapia personalizzata col test RAS
- Esofagite: cause, cronica, sintomi, cura, dieta, alimentazione
- Esofagite da reflusso: cause e fattori di rischio
- Esofagite da reflusso: sintomi e segni
- Esofagite da reflusso: esami, diagnosi, endoscopia, breath test
- Esofagite da reflusso: gravità e complicanze
- Esofagite da reflusso: terapia, farmaci, chirurgia
- Esofagite da reflusso: consigli, dieta, cibi da evitare, come dormire, prevenzione
- Reflusso gastro-esofageo nell’anziano: epidemiologia, cause, fattori di rischio
- Reflusso gastro-esofageo nell’anziano: sintomi e segni
- Reflusso gastro-esofageo nell’anziano: esami, diagnosi, gravità
- Reflusso gastro-esofageo nell’anziano: complicanze, prevenzione, dieta
- Reflusso gastro-esofageo nell’anziano: terapia a breve termine, farmaci, interazioni
- Reflusso gastro-esofageo nell’anziano: terapia a lungo termine, chirurgia, farmaci
Lo Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o unisciti al nostro gruppo Facebook o ancora seguici su Twitter, su Instagram o su Pinterest, grazie!
