
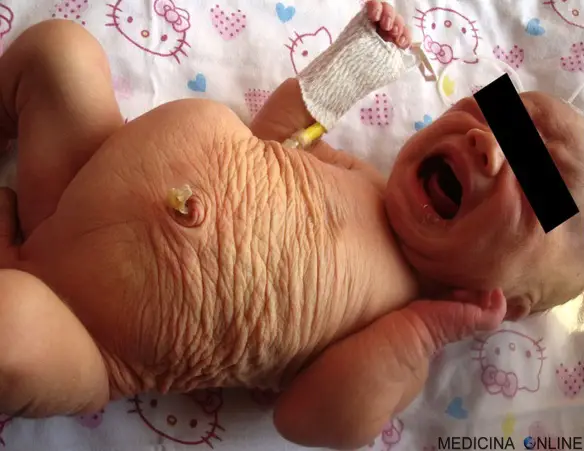
 La “sindrome dell’addome a prugna secca” (anche chiamata “sindrome della pancia a prugna secca” o “sindrome della pancia a prugna” o “sindrome di Eagle-Barret” o “sindrome di Obrinsky“ o “sindrome da deficit dei muscoli addominali” o “sindrome della triade“; in inglese “prune belly syndrome“) è una sindrome rara, congenita (già presente alla nascita) e ad eziologia sconosciuta (non si conoscono le cause). Il nome di questa sindrome deriva dal caratteristico aspetto grinzoso della pelle della parete addominale nei neonati, che ricorda appunto quello di un frutto secco (prune belly significa pancia a prugna).
La “sindrome dell’addome a prugna secca” (anche chiamata “sindrome della pancia a prugna secca” o “sindrome della pancia a prugna” o “sindrome di Eagle-Barret” o “sindrome di Obrinsky“ o “sindrome da deficit dei muscoli addominali” o “sindrome della triade“; in inglese “prune belly syndrome“) è una sindrome rara, congenita (già presente alla nascita) e ad eziologia sconosciuta (non si conoscono le cause). Il nome di questa sindrome deriva dal caratteristico aspetto grinzoso della pelle della parete addominale nei neonati, che ricorda appunto quello di un frutto secco (prune belly significa pancia a prugna).
Epidemiologia
L’incidenza annuale è stimata in 1/35.000 – 1/50.000. Nel 95% dei casi la malattia colpisce i maschi.
Cause e fattori di rischio
L’eziologia non è nota. Si ritiene che la malattia origini dall’ostruzione dell’uretra nelle prime fasi dello sviluppo; ciò causa una notevole distensione della vescica e asciti urinarie che esitano nell’iposviluppo della muscolatura della parete addominale e nella mancata discesa dei testicoli. La mancata eliminazione dell’urina dalla vescica causa inoltre oligoidramnios che, a sua volta, compromette lo sviluppo dei polmoni, esitando nei segni clinici caratteristici della sequenza di Potter. L’ostruzione congenita delle vie urinarie in una fase critica dell’organogenesi può avere conseguenze gravi e permanenti sulla funzione dei reni, dell’uretere e della vescica. È stata ipotizzata un’origine genetica o un mosaicismo nei casi di ricorrenza della sindrome ”prune belly” tra i fratelli, anche se la maggior parte dei casi è sporadica.
Leggi anche:
- Differenza tra autosomica dominante e recessiva con esempi
- Ereditarietà X linked, malattie dominanti e recessive legate al cromosoma X: significato, esempi
Sintomi e segni
La sindrome prune belly è caratterizzata da:
- oligoidramnios,
- vescica molto grande e distesa,
- idroureteronefrosi bilaterale grave,
- diastasi addominale,
- ascite fetale,
- displasia renale,
- pervietà dell’uraco,
- criptorchidismo,
- ipoplasia dei polmoni,
- piede equino.
Possono essere presenti i segni clinici caratteristici della sequenza di Potter, che comprendono:
- occhi iperdistanziati,
- naso appiattito,
- mento arretrato,
- orecchie grandi,
- basso impianto di cartilagini.
La malattia si associa inoltre a :
- difetti polmonari,
- difetti scheletrici,
- difetti cardiaci,
- difetti gastrointestinali,
- atresia dell’uretra.
I neonati presentano un addome grinzoso (”a prugna secca”), che successivamente assume l’aspetto di ”addome pendulo”. I bambini sono predisposti alle infezioni delle vie urinarie secondarie allo svuotamento incompleto della vescica.
Diagnosi
La sindrome prune belly spesso viene diagnosticata in epoca prenatale nel corso di ecografie routinarie. La diagnosi postnatale si basa sui segni clinici caratteristici, sulla valutazione della funzione polmonare, sull’ecografia e sul cistouretrogramma da svuotamento. La diagnosi differenziale in utero si pone con la megacisti/megauretere e con le valvole uretrali posteriori. La diagnosi prenatale si basa sull’esame ecografico. Analogamente a tutte le altre forme di LUTO, la consulenza è incentrata sulle conseguenze postnatali, che comprendono la grave ipoplasia polmonare secondaria all’oligoidramnios, la morte in utero e i segni clinici caratteristici della sindrome in assenza di compromissione renale. La consulenza genetica può essere utile qualora un fratello sia affetto. Sono stati descritti casi di trasmissione autosomica dominante e legata all’X. La maggior parte dei casi è sporadica, anche se sono stati descritti casi di ricorrenza tra fratelli.
Terapia
Durante la gravidanza, è necessario monitorare le vie urinarie e il volume del liquido amniotico mediante ecografia. Si raccomanda la decompressione precoce delle ostruzioni gravi che contribuiscono all’insorgenza dell’oligoidramnios, mediante lo svuotamento vescicale. La profilassi antibiotica deve essere avviata alla nascita. Altri trattamenti comprendono l’orchidopessia bilaterale e l’addominoplastica, oltre ad eventuali interventi chirurgici di ricostruzione urologica.
Prognosi
La prognosi dipende dal grado di dilatazione delle vie urinarie e dalla gravità della compromissione della funzione renale. L’esito è favorevole quando il volume del liquido amniotico è normale. La grave ostruzione delle vie urinarie, come nel caso dell’atresia uretrale, esita nell’oligoidramnios progressivo, nell’ipoplasia polmonare e nella morte nel periodo neonatale. La causa più comune di morte postnatale è l’urosepsi. Tuttavia, sono noti casi di bambini che presentano crescita e funzione renale normali anche in presenza di significative anomalie delle vie urinarie documentate dalle radiografie.
Leggi anche:
- Diastasi addominale in gravidanza, parto, neonati: cause, sintomi, terapie
- Diastasi addominale: cos’è e cosa la causa? Si può curare con fisioterapia ed esercizi fisici?
- Diastasi della sinfisi pubica: cause, sintomi, diagnosi e terapie
- Sindrome dell’addome a prugna secca: cause, sintomi, diagnosi, terapie
- Addominoplastica: rischi, costi, cicatrice, convalescenza
- Diastasi in medicina: significato, esempi, tipi principali
- Quanto è normale ingrassare in gravidanza?
- Come fare gli addominali alti, bassi e laterali (obliqui), consigli per livello base ed avanzato, come evitare i dieci errori più comuni [VIDEO]
- Sono normopeso, sottopeso o sovrappeso? Come si calcola l’Indice di Massa Corporea (BMI)?
- Ascite: cura, addominale, tumore, sintomi iniziali, paracentesi
- Differenza tra ernia e laparocele
- Differenza tra laparotomia e laparoscopia
- Nuova gravidanza: meglio se tra un anno ed un anno e mezzo di distanza dalla prima
- Ernia interna ed esterna: definizione, significato, classificazione
- Ernia inguinale: rimedi, intervento, donne, uomini, strozzata, cura
- Ernia crurale: strozzata, anatomia, intervento, convalescenza
- Ernia iatale: rimedi, dieta, farmaci, da scivolamento, dolore, diagnosi
- Ernia ombelicale: intervento, quando operare, convalescenza
- Ernia epigastrica: cicatrice, convalescenza, recidiva
- Quadranti addominali: semeiotica, anatomia ed organi contenuti
- Regioni addominali: semeiotica, anatomia ed organi contenuti
- Laparoscopia addominale, ginecologica, anestesia, rischi, convalescenza
- Laparotomia addominale, ginecologica, anestesia, rischi, convalescenza
- Laparocele: sintomi, strozzato, rischi, intervento, quando intervenire
- Cos’è la sinfisi pubica: anatomia, dove si trova, a che serve
- Differenze tra sinfisi pubica maschile e femminile e come cambia in gravidanza e parto
- Differenza tra bacino, pelvi, cresta iliaca, coccige, osso sacro, ileo, sinfisi pubica e ischio
- Aborto spontaneo: quali sono le cause ed i sintomi precoci?
- Aborto spontaneo: cos’è, sintomi iniziali e come riconoscerlo
- Perdite bianche, gialle marroni in gravidanza: quando preoccuparsi e cosa fare?
- Rimanere incinta: i 30 migliori consigli alla coppia per aumentare le possibilità di gravidanza
- Parto pretermine: conseguenze sul neonato prematuro
- Dolori del parto: epidurale, controllo autonomo e medico
- Differenza tra parto naturale, indotto e pilotato
- Dopo il parto: come recuperare l’intimità di coppia?
- Morte in culla (SIDS): prevenzione, cause, sintomi e percentuale dei casi
- Parto prematuro: cause, fattori di rischio, prevenzione e psicologia dei genitori
- In quale settimana di parto un neonato viene detto prematuro?
- Integratori per la fertilità che aumentano le possibilità di gravidanza
- Percentuale di sopravvivenza del neonato prematuro: peso e prognosi
- Incubatrice neonatale: funzionamento, prezzo, per quali neonati si usa?
- Differenza tra culla termica ed incubatrice neonatale
- Cerco la gravidanza: quanto tempo è necessario per rimanere incinta?
- New York legalizza l’aborto fino al nono mese di gravidanza
- Differenza tra embrione e feto
- La gravidanza extrauterina: come riconoscere i sintomi ed intervenire in tempo
- Raschiamento: è doloroso, quando avere rapporti, perdite e nuova gravidanza
- Aborto spontaneo: quali sono le cause ed i sintomi precoci?
- Aborto spontaneo: cos’è, sintomi iniziali e come riconoscerlo
- Per abortire serve il consenso dei genitori? Aborto per minorenni e interdette
- Aborto: entro quanto è legale?
- Aborto volontario: decide la madre, il padre o entrambi? E se la donna è minorenne?
- Dove abortire? Il medico è costretto a praticare l’aborto? Cos’è l’obiezione di coscienza?
- Differenza tra aborto spontaneo, completo, incompleto, ritenuto
- Differenza tra aborto spontaneo, parto prematuro e “nato morto”
- Differenza tra aborto interno e spontaneo
- Differenza tra aborto spontaneo, provocato, volontario e terapeutico
- Differenza tra aborto e interruzione di gravidanza
- Differenza tra aborto e morte intrauterina fetale
- Differenza tra aborto e gravidanza biochimica
- Sifilide in gravidanza: conseguenze, sintomi nei neonati, diagnosi, cure
- Qual è il mese migliore per partorire per te e per il bambino?
- Ipoposia: quando lo sperma è troppo poco. Cause e terapie per aumentare la quantità di eiaculato
- Isteroscopia: preparazione, è dolorosa, polipo, quando farla, costo
- Isterosalpingografia: come si fa, dolore, preparazione, rischi, costo
- Idrosalpinge: cos’è, ecografia, cura, gravidanza, intervento
- Tube di Falloppio chiuse: cosa sono e come “aprirle”?
- Salpingi (tube di Falloppio): cosa sono, dove sono ed a che servono?
- Differenza tra isterosalpingografia e isteroscopia
- Differenza tra isteroscopia e colposcopia
- Differenza tra parto eutocico e spontaneo
- Differenza tra parto eutocico (fisiologico) e distocico
- Differenza tra parto a termine, pretermine, abortivo e post-termine
- Macrosomia, distocico, episiotomia, indice di Bishop… Il vocabolario del parto
- Travaglio, secondamento e le altre fasi del parto naturale
- Differenza tra presentazione cefalica, podalica e trasversale nel parto
- Perché riprendere l’attività sessuale dopo il parto è così difficile?
- Tumore dell’endometrio: sopravvivenza, metastasi, si guarisce?
- Sindrome da iperstimolazione ovarica dopo pick up
- Endometriosi: cause, sintomi e menopausa
- Differenza placenta bassa e previa: rischi e cosa evitare
- Posso rimanere incinta a 50 anni?
- Problemi a rimanere incinta? Rimedi naturali per diventare mamma
- Acido folico (vitamina B9): a cosa serve, in quali alimenti trovarlo e perché è importante PRIMA e durante la gravidanza
- Salmonella in gravidanza: trasmissione, rischi, consigli per evitarla
- Ho preso la salmonella in gravidanza: quali danni al feto?
- Differenze tra bambini nati con cesareo e quelli con parto naturale
- Le paure del parto cesareo e di quello naturale
- L’allattamento al seno è possibile dopo un parto cesareo?
- Differenze tra neonati allattati con latte materno ed artificiale
- Parto cesareo programmato: cosa mi succederà dopo?
- Diarrea in gravidanza: cosa mangiare, cosa fare e rimedi
- Differenza tra infertilità e sterilità
- Differenza tra sterilità primaria e secondaria
- Non riesco a rimanere incinta: colpa dell’utero
- Non riesco a rimanere incinta: e se la colpa fosse dell’uomo?
- Differenza tra infertilità e impotenza
- New York legalizza l’aborto fino al nono mese di gravidanza
- Non riesco a rimanere incinta: colpa di una infezione ginecologica
- Non riesco a rimanere incinta: colpa delle ovaie che non funzionano bene
- Pubalgia in gravidanza: cause e rimedi del dolore all’osso pubico
- Differenza tra ovaio microcistico, policistico e cisti ovariche
- Differenza tra ovaio policistico, micropolicistico e microfollicolare
- Differenza tra ovaio policistico e multifollicolare
- Ovaio: anatomia, funzioni e patologie in sintesi
- Vagina: anatomia, funzioni e patologie in sintesi
- Come e quando fare il test di gravidanza
- Differenza dei capezzoli e del seno in gravidanza
- Fino a che età una donna può avere figli?
- Differenza tra vagina e vulva
- Utero: anatomia, funzioni, patologie e sintomi in sintesi
- Apparato genitale femminile: anatomia, funzioni e patologie in sintesi
- La vagina è uguale in tutte le donne?
- Quanto è profonda una vagina?
Dott. Emilio Alessio Loiacono
Medico Chirurgo
Direttore dello Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o unisciti al nostro gruppo Facebook o ancora seguici su Twitter, su Instagram, su YouTube, su LinkedIn e su Pinterest, grazie!
