
 La biodisponibilità in farmacologia è il grado (e talvolta velocità) nel quale la forma attiva di un farmaco (cioè il farmaco stesso o un suo metabolita) raggiunge la circolazione sistemica, acquisendo così la capacità di accedere al suo sito di azione. Le proprietà fisico-chimiche di un farmaco sono responsabili del suo potenziale di assorbimento, ma le proprietà della forma farmaceutica (che in parte dipendono dalla sua progettazione e fabbricazione) possono determinare in larga misura la sua biodisponibilità. Le differenze di biodisponibilità tra le formulazioni di un determinato farmaco possono avere un’importanza clinica non trascurabile. Di conseguenza, il concetto di equivalenza tra le varie preparazioni farmaceutiche è determinante per poter prendere decisioni cliniche avvedute. L’equivalenza chimica si riferisce alle preparazioni farmaceutiche che contengono lo stesso composto nella medesima quantità e che soddisfano gli standard ufficiali vigenti; tuttavia, i componenti farmacologicamente inattivi presenti nelle preparazioni possono essere diversi. La bioequivalenza si riferisce agli equivalenti chimici che, quando vengono somministrati alla stessa persona con il medesimo regime di dosaggio, danno luogo a concentrazioni equivalenti del farmaco nel sangue e nei tessuti. L’equivalenza terapeutica si riferisce alle preparazioni farmaceutiche che, quando vengono somministrate alla stessa persona con il medesimo regime di dosaggio, danno origine essenzialmente allo stesso effetto terapeutico o alla stessa tossicità. È logico attendersi che le preparazioni bioequivalenti siano anche terapeuticamente equivalenti.
La biodisponibilità in farmacologia è il grado (e talvolta velocità) nel quale la forma attiva di un farmaco (cioè il farmaco stesso o un suo metabolita) raggiunge la circolazione sistemica, acquisendo così la capacità di accedere al suo sito di azione. Le proprietà fisico-chimiche di un farmaco sono responsabili del suo potenziale di assorbimento, ma le proprietà della forma farmaceutica (che in parte dipendono dalla sua progettazione e fabbricazione) possono determinare in larga misura la sua biodisponibilità. Le differenze di biodisponibilità tra le formulazioni di un determinato farmaco possono avere un’importanza clinica non trascurabile. Di conseguenza, il concetto di equivalenza tra le varie preparazioni farmaceutiche è determinante per poter prendere decisioni cliniche avvedute. L’equivalenza chimica si riferisce alle preparazioni farmaceutiche che contengono lo stesso composto nella medesima quantità e che soddisfano gli standard ufficiali vigenti; tuttavia, i componenti farmacologicamente inattivi presenti nelle preparazioni possono essere diversi. La bioequivalenza si riferisce agli equivalenti chimici che, quando vengono somministrati alla stessa persona con il medesimo regime di dosaggio, danno luogo a concentrazioni equivalenti del farmaco nel sangue e nei tessuti. L’equivalenza terapeutica si riferisce alle preparazioni farmaceutiche che, quando vengono somministrate alla stessa persona con il medesimo regime di dosaggio, danno origine essenzialmente allo stesso effetto terapeutico o alla stessa tossicità. È logico attendersi che le preparazioni bioequivalenti siano anche terapeuticamente equivalenti.
I problemi terapeutici (p. es., tossicità, mancanza di efficacia) si incontrano più frequentemente nel corso dei trattamenti di lunga durata quando a un paziente ormai stabilizzato con l’impiego di una certa formulazione viene somministrato un farmaco non equivalente in sostituzione del primo (come avviene per la digossina o la fenitoina).
Talvolta l’equivalenza terapeutica può essere ottenuta nonostante le differenze di biodisponibilità. Per esempio, l’indice terapeutico (rapporto tra la massima dose tollerata e la minima dose efficace) della penicillina è talmente ampio che discrete differenze di concentrazione ematica dovute alle differenze di biodisponibilità tra le varie preparazioni penicilliniche possono non influenzare l’efficacia o la sicurezza terapeutica. Al contrario, le differenze di biodisponibilità sono importanti per un farmaco con un indice terapeutico relativamente ristretto.
La biodisponibilità è influenzata anche dalle caratteristiche fisiologiche del paziente e dalla presenza di patologie concomitanti. La velocità di assorbimento è importante, perché anche quando un farmaco viene assorbito completamente, esso può essere assorbito troppo lentamente per produrre con sufficiente rapidità una concentrazione terapeutica nel sangue, oppure così velocemente da causare tossicità per le elevate concentrazioni raggiunte dopo ogni dose.
Cause di bassa biodisponibilità
Quando un farmaco si dissolve rapidamente e attraversa facilmente le membrane, l’assorbimento tende a essere completo, ma l’assorbimento dei farmaci somministrati per via orale non è sempre completo. Prima di raggiungere la vena cava, un farmaco deve percorrere il canale GI e passare attraverso la parete intestinale e il fegato, sedi comuni di metabolizzazione dei farmaci; pertanto, un farmaco può essere metabolizzato (metabolismo di primo passaggio) prima ancora di poter essere dosato nella circolazione sistemica. Molti farmaci hanno una bassa biodisponibilità per via orale a causa del cospicuo metabolismo di primo passaggio. Per tali farmaci (p. es., l’isoproterenolo, la noradrenalina, il testosterone), l’estrazione a livello di questi tessuti è così ampia che la biodisponibilità è praticamente nulla. Per i farmaci che possiedono un metabolita attivo, le conseguenze terapeutiche del metabolismo di primo passaggio dipendono dal contributo relativo del farmaco e del metabolita agli effetti desiderati e indesiderati.
Una bassa biodisponibilità si osserva più comunemente con le preparazioni orali dei farmaci poco idrosolubili che vengono assorbiti lentamente. Quando l’assorbimento è lento o incompleto, la biodisponibilità può essere influenzata da un maggior numero di fattori rispetto a quanto avviene con un assorbimento rapido e completo; in questo modo, un assorbimento incompleto o lento conduce spesso a risposte terapeutiche variabili.
La permanenza nel tratto GI per un tempo insufficiente è una causa frequente di bassa biodisponibilità. I farmaci assunti per via orale rimangono a contatto con la parete dell’intero tratto GI per non più di 1 o 2 gg e con quella dell’intestino tenue solamente per 2-4 h. Se il farmaco non si dissolve facilmente o non è in grado di attraversare efficacemente la membrana epiteliale (p. es., se è altamente ionizzato e polare), il tempo di permanenza a livello della sede di assorbimento può non essere sufficiente. In queste circostanze la biodisponibilità, oltre a essere bassa, tende a subire variazioni considerevoli. L’età, il sesso, l’attività fisica, il fenotipo genetico, lo stress, le malattie (p. es., l’acloridria, le sindromi da malassorbimento) o precedenti interventi chirurgici sull’apparato GI possono influenzare la biodisponibilità dei farmaci.
Essa inoltre può essere ridotta dalle reazioni chimiche che entrano in competizione con l’assorbimento. Queste reazioni includono la formazione di complessi (p. es., fra la tetraciclina e gli ioni metallici polivalenti), l’idrolisi per opera del succo gastrico acido o degli enzimi digestivi (p. es., l’idrolisi della penicillina e del cloramfenicolo palmitato), la coniugazione a livello della parete intestinale (p. es., la coniugazione con zolfo dell’isoproterenolo), l’adsorbimento ad altri farmaci (p. es., la digossina e la colestiramina) e il metabolismo da parte della microflora intestinale.
Valutazione della biodisponibilità
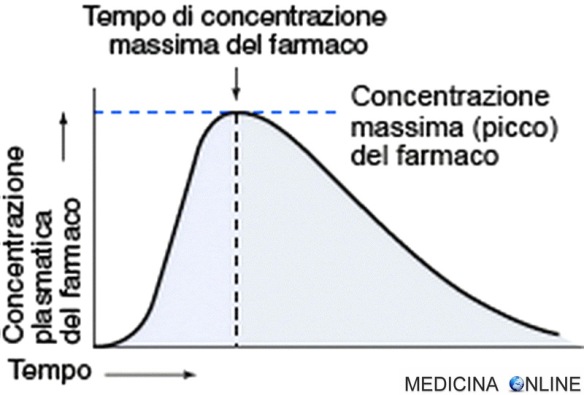
La valutazione della biodisponibilità effettuata mediante le misurazioni seriate della concentrazione plasmatica comporta solitamente la determinazione della concentrazione plasmatica massima (di picco) del farmaco, quella del tempo necessar
io per raggiungere la concentrazione plasmatica massima (tempo di picco) e il calcolo dell’area al di sotto della curva concentrazione plasmatica-tempo (Area Under plasma concentration-time Curve, AUC, v. Fig. 298-1). La concentrazione plasmatica dei farmaci aumenta con l’entità dell’assorbimento; il picco viene raggiunto quando la velocità di eliminazione del farmaco diviene uguale alla velocità di assorbimento. Le determinazioni della biodisponibilità basate sulla sola concentrazione plasmatica di picco possono essere ingannevoli, perché l’eliminazione dei farmaci ha inizio appena essi entrano in circolo. L’indice generico della velocità di assorbimento utilizzato più diffusamente è il tempo di picco; più è lento l’assorbimento, più il tempo di picco è tardivo. Tuttavia spesso il tempo di picco non rappresenta una buona misura statistica, perché è un parametro di tipo discreto che dipende dalla frequenza con cui vengono prelevati i campioni di sangue e, nel caso di curve di concentrazione relativamente piatte in prossimità del picco, dalla riproducibilità dell’analisi.
L’AUC è la misura più attendibile della biodisponibilità. Essa è direttamente proporzionale alla quantità totale di farmaco immodificato che raggiunge la circolazione sistemica. Per una determinazione accurata, il sangue deve essere prelevato frequentemente per un periodo di tempo abbastanza lungo da osservare l’eliminazione pressoché completa del farmaco. Le preparazioni farmaceutiche possono essere considerate bioequivalenti per grado e velocità di assorbimento se le loro curve di concentrazione plasmatica sono sostanzialmente sovrapponibili. Le preparazioni che possiedono AUC simili ma le cui curve di concentrazione plasmatica hanno un andamento differente sono equivalenti per il grado di assorbimento, ma differiscono quanto al profilo velocità-tempo di assorbimento.
Dose singola o dosi multiple: la biodisponibilità può essere valutata dopo una dose singola oppure dopo dosi ripetute (multiple). Dopo una dose singola si ottengono più informazioni sulla velocità di assorbimento di quanto non avvenga dopo somministrazioni multiple. Tuttavia, queste ultime rappresentano più da vicino le circostanze cliniche abituali e inoltre le concentrazioni plasmatiche sono solitamente più elevate rispetto a quelle che si osservano dopo una dose singola, facilitando l’analisi dei dati. Dopo più somministrazioni separate da un intervallo di tempo prefissato per un periodo pari a quattro o cinque volte l’emivita di eliminazione, la concentrazione ematica del farmaco dovrebbe trovarsi allo stato di equilibrio (cioè la quantità assorbita equivale alla quantità eliminata in ogni intervallo di somministrazione). L’entità dell’assorbimento può quindi essere analizzata misurando l’AUC in corrispondenza di un intervallo di somministrazione. La misurazione dell’AUC nelle 24 h è probabilmente da preferire, a causa delle variazioni circadiane delle funzioni fisiologiche e delle possibili variazioni degli intervalli di somministrazione e delle velocità di assorbimento durante la giornata.
Per i farmaci escreti principalmente immodificati con le urine, la biodisponibilità può essere stimata misurando la quantità totale del farmaco escreta dopo una singola somministrazione. Idealmente, le urine vengono raccolte per un periodo pari a 7-10 volte l’emivita di eliminazione, in modo da ritrovarvi tutto il farmaco assorbito. La biodisponibilità può essere determinata anche dopo somministrazioni multiple mediante la determinazione del farmaco immodificato presente nelle urine delle 24 h in condizioni stazionarie.
Per approfondire:
- Assorbimento dei farmaci: somministrazione orale, parenterale e forme a rilascio controllato
- Diffusione attiva, passiva o pinocitosi: il trasporto dei farmaci attraverso le membrane cellulari
- Distribuzione, velocità di ingresso, equilibrio di distribuzione e legame di un farmaco
- Finestra terapeutica ed esempi di farmaci con ampi e ristretti indici terapeutici
- Eliminazione di un farmaco: metabolismo, citocromo P-450, coniugazione
- Eliminazione di un farmaco: escrezione renale e biliare
- Farmacocinetica, biodisponibilità, volume di distribuzione, emivita di un farmaco e variabilità individuale
- Farmacodinamica, interazioni farmaco-recettore e relazione dose-risposta
- Farmacogenetica: variabilità farmacocinetica e farmacodinamica
- Interazioni farmacologiche: farmacodinamiche e farmacocinetiche
- Reazioni avverse ai farmaci: effetti collaterali, tossicità, allergie, idiosincrasie
- Rapporto rischi-benefici nell’assunzione dei farmaci
- Mancata compliance del paziente: quando il paziente non prende i suoi farmaci
Leggi anche:
- Differenza tra farmacodinamica e farmacocinetica
- Il paracetamolo (Tachipirina) è un antinfiammatorio non steroideo (FANS)?
- Antinfiammatori non steroidei (FANS): significato ed elenco di farmaci
- Differenze tra Paracetamolo, Tachipirina, Ibuprofene, Aspirina, Efferalgan e Co-Efferalgan
- Comportamento dei farmaci nella barriera emato-encefalica
- Meglio Aspirina o Ibuprofene?
- A che serve la Tachipirina (paracetamolo)?
- Differenza tra farmaco originale, generico ed equivalente
- Che significa farmaco non steroideo?
- Che significa il termine “farmaco”: definizione ed etimologia
- Differenza tra farmaci di fascia A, B, C, H
- Differenza tra farmaco etico, da banco, senza obbligo di ricetta, speciale e limitativa
- Differenza tra farmaco a rilascio prolungato, ritardato, modificato, ripetuto, controllato
- Che significa farmaco di fascia C o Classe C?
- Che significa farmaco etico, esempio, classe A, è detraibile, elenco
- Farmaco “ideale”: quali sono le sue caratteristiche?
- Che significa farmaco da banco?
- Differenza tra farmaco etico e generico
- Che significa farmaco mutuabile?
- Che significa farmaco ripetibile? Caratteristiche della ricetta
- Cos’è un farmaco biologico, a che serve e come funziona?
- Che significa farmaco generico o equivalente?
- Antiossidanti: alimenti ed integratori migliori contro i radicali liberi
- Sali minerali: definizione, funzioni, alimenti, integratori [GUIDA COMPLETA]
- Differenza tra omeopatia, fitoterapia ed erboristeria
- Differenza tra Eutirox e Ibsa nella cura dell’ipotiroidismo
- Ipertensione: quali farmaci usare per abbassare la pressione arteriosa?
- Coumadin: quando si usa, dosaggio ed effetti collaterali (foglio illustrativo)
- Cos’è una sostanza stupefacente?
- Gli integratori alimentari sono doping? Elenco delle sostanze dopanti
- Differenza tra integratori alimentari e sostanze dopanti con esempi
- Differenze tra effetto collaterale, effetti indesiderati, reazione avversa, evento avverso
- Differenza tra controindicazioni ed effetti indesiderati
- Che significa “ai pasti”? Quando assumere i farmaci?
- Che significa “effetto placebo” e perché un placebo funziona?
- Floriterapia e Fiori di Bach: elenco, classificazione, funzioni
- Omeopatia: cos’è? I farmaci omeopatici funzionano realmente?
- Stitichezza o stipsi acuta e cronica: terapie farmacologiche
- Reflusso gastroesofageo: terapia farmacologica e chirurgica
- I migliori farmaci antiacidi da banco, senza ricetta medica
- Eutirox può essere usato anche per dimagrire? Quali sono i rischi?
- Eutirox: effetti collaterali e controindicazioni del farmaco
- Come fare un clistere evacuativo: procedura semplice con enteroclisma
- Fare un clistere evacuativo: procedura semplice con peretta
- Microclisma: cos’è e come si usa in adulti e neonati
- Differenze tra clistere, peretta, enteroclisma, microclisma
- Guida facile per fare una iniezione intramuscolare corretta ed indolore
- In quali parti del corpo e dove si esegue una iniezione intramuscolare?
- Com’è fatta una siringa e come si usa correttamente?
- Differenza tra dolore acuto, cronico, persistente ed episodico con esempi
- Differenza tra dolore somatico e psicosomatico
- Dolore: come si misura? La scala visiva e numerica verbale
- Dolore: quando chiamare il medico e cosa riferirgli
- Dolore: esistono esami specifici per rilevarlo?
- Meglio Aspirina o Ibuprofene?
- Supposta rettale: vantaggi e svantaggi rispetto ad altre vie di somministrazione
- Cosa succede ad una supposta dopo averla inserita? Come funziona?
- Supposte di glicerina: come usarle in bambini, adulti, gravidanza
- Come mettere facilmente una supposta a neonati, bambini, adulti
- Si possono tagliare o spezzare le supposte rettali?
- Lingua bianca, impastata, spaccata: cause e quando è pericolosa
- Vie di somministrazione di un farmaco: tipi, differenze, vantaggi e svantaggi
- A che serve la Tachipirina (paracetamolo)?
- Gravidanza e allattamento: posso assumere Tachipirina, Ibuprofene e Co-Efferalgan? Quante compresse?
- Differenze tra Efferalgan e Co-Efferalgan
- Cardioaspirin 100mg: effetti indesiderati, a cosa serve, dosaggi (foglio illustrativo)
- Che significa somministrazione di un farmaco PER OS o PO?
- Via di somministrazione orale, per os: vantaggi e svantaggi
- Tachipirina, paracetamolo, Efferalgan: posologia, controindicazioni ed effetti collaterali
- Voltaren Emulgel (diclofenac): come usarlo, gravidanza ed effetti collaterali
- Arvenum: terapia di emorroidi e fragilità capillari
- Enterogermina per gonfiore, diarrea e dolori addominali: foglietto illustrativo
- Moment (ibuprofene): posologia, effetti collaterali, gravidanza, prezzo
- Rinazina spray nasale in bambini e adulti: posologia e prezzo
- Glicerolo Carlo Erba soluzione rettale e supposte: posologia ed effetti collaterali
- Triatec (ramipril): posologia, effetti collaterali [FOGLIETTO ILLUSTRATIVO]
- Lasix (furosemide): posologia, effetti collaterali [FOGLIETTO ILLUSTRATIVO]
- Norvasc (amlodipina): posologia, effetti collaterali [FOGLIETTO ILLUSTRATIVO]
- Augmentin (amoxicillina): posologia, effetti collaterali [FOGLIETTO ILLUSTRATIVO]
- Omeprazen (omeprazolo): posologia, effetti collaterali [FOGLIETTO ILLUSTRATIVO]
- Dibase (vitamina D): posologia, effetti collaterali [FOGLIETTO ILLUSTRATIVO]
- Torvast (atorvastatina): posologia, effetti collaterali [FOGLIETTO ILLUSTRATIVO]
- Differenza tra Gabapentin e Pregabalin
- Farmaco Lyrica (Pregabalin): indicazioni ed effetti collaterali
- Lyrica (Pregabalin): è un farmaco che fa ingrassare?
Lo Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o unisciti al nostro gruppo Facebook o ancora seguici su Twitter, su Instagram o su Pinterest, grazie!
