
 L’assorbimento dei farmaci è determinato dalle loro proprietà fisico-chimiche, dalle loro formulazioni e dalle vie di somministrazione. I prodotti farmaceutici, cioè le effettive preparazioni (p. es., compresse, capsule, soluzioni) costituite dal farmaco e dagli eccipienti, sono formulate per essere somministrate per varie vie, tra le quali l’orale, la buccale, la sub-linguale, la rettale, la parenterale, la topica e l’inalatoria. Un requisito essenziale per l’assorbimento è la dissoluzione del farmaco. I prodotti farmaceutici solidi (p. es., le compresse) si disintegrano e si disgregano, ma l’assorbimento può avvenire solo dopo che i farmaci sono entrati in soluzione.
L’assorbimento dei farmaci è determinato dalle loro proprietà fisico-chimiche, dalle loro formulazioni e dalle vie di somministrazione. I prodotti farmaceutici, cioè le effettive preparazioni (p. es., compresse, capsule, soluzioni) costituite dal farmaco e dagli eccipienti, sono formulate per essere somministrate per varie vie, tra le quali l’orale, la buccale, la sub-linguale, la rettale, la parenterale, la topica e l’inalatoria. Un requisito essenziale per l’assorbimento è la dissoluzione del farmaco. I prodotti farmaceutici solidi (p. es., le compresse) si disintegrano e si disgregano, ma l’assorbimento può avvenire solo dopo che i farmaci sono entrati in soluzione.
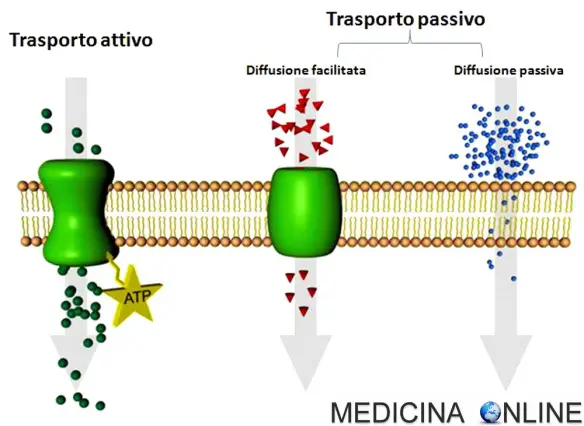
Trasporto attraverso le membrane cellulari
La maggior parte delle vie di somministrazione (esclusa quella EV) implica la necessità che i farmaci attraversino diverse membrane cellulari semipermeabili prima di raggiungere la circolazione sistemica. Queste membrane sono barriere biologiche che impediscono in maniera selettiva il passaggio delle molecole dei farmaci e sono composte principalmente da una matrice molecolare lipidica bistratificata, contenente soprattutto colesterolo e fosfolipidi. I lipidi conferiscono stabilità alla membrana e sono responsabili delle sue caratteristiche di permeabilità. Nello spessore della matrice lipidica sono inserite proteine globulari di diverse dimensioni e composizione, le quali sono coinvolte nei processi di trasporto e funzionano come recettori per la regolazione delle attività cellulari. I farmaci possono attraversare una barriera biologica mediante i meccanismi della diffusione passiva, della diffusione passiva facilitata, del trasporto attivo o della pinocitosi.
Leggi anche:
- Differenza tra cellula aploide e diploide con esempi
- Differenza tra cellule eucariote e procariote
- Riproduzione cellulare e ciclo cellulare
- Mitosi: spiegazione delle quattro fasi
- Meiosi: spiegazione di tutte tappe
Diffusione passiva
In questo processo, il trasporto di un soluto attraverso una membrana cellulare dipende dal suo gradiente di concentrazione. La maggior parte delle molecole dei farmaci attraversa le membrane per diffusione semplice da una regione ad alta concentrazione (p. es., i fluidi GI) a una regione a bassa concentrazione (p. es., il sangue). Poiché le molecole dei farmaci vengono rapidamente rimosse per opera del torrente circolatorio e distribuite in un ampio volume di liquidi e tessuti dell’organismo, la loro concentrazione nel sangue è inizialmente bassa rispetto a quella presente nella sede di somministrazione, dando luogo a un gradiente elevato. La velocità di diffusione è direttamente proporzionale al gradiente, ma dipende anche dalla liposolubilità, dal grado di ionizzazione e dalle dimensioni della molecola, nonché dall’area della superficie di assorbimento. Dal momento che la membrana cellulare è di natura lipidica, i farmaci liposolubili diffondono più velocemente di quelli relativamente non liposolubili. Le molecole di piccole dimensioni tendono a passare attraverso le membrane più rapidamente di quelle voluminose.
La maggior parte dei farmaci è rappresentata da basi o acidi organici deboli che in ambiente acquoso si trovano in forma ionizzata e in forma non ionizzata. La forma non ionizzata di solito è liposolubile e diffonde facilmente attraverso le membrane cellulari; la forma ionizzata non è in grado di attraversare con facilità la membrana cellulare a causa della sua bassa liposolubilità e della sua alta resistenza elettrica, derivante dalla carica della molecola e dai gruppi polari presenti sulla superficie della membrana stessa. Di conseguenza, la penetrazione dei farmaci nei compartimenti biologici può essere attribuita per lo più alla loro forma non ionizzata. In condizioni di equilibrio, la distribuzione di un farmaco ionizzabile sui due versanti di una membrana è determinata dal pKa del farmaco (il pH al quale le concentrazioni della sua forma non ionizzata e di quella ionizzata sono uguali) e dal gradiente di pH, qualora sia presente. Per un acido debole, più elevato è il pH, più basso è il rapporto tra la forma non ionizzata e quella ionizzata. Nel plasma (pH 7,4), il rapporto tra la forma non ionizzata e quella ionizzata di un acido debole (p. es., con pKa di 4,4) è di 1:1000; nel succo gastrico (pH 1,4) il rapporto è invertito (1000:1). Quando l’acido debole viene somministrato per via orale, il gradiente di concentrazione del farmaco non ionizzato tra lo stomaco e il plasma tende a essere elevato, favorendone la diffusione attraverso la mucosa gastrica. In condizioni di equilibrio, le concentrazioni del farmaco non ionizzato nello stomaco e nel plasma sono uguali, perché solo il farmaco non ionizzato può passare attraverso le membrane; la concentrazione del farmaco ionizzato nel plasma sarebbe quindi circa 1000 volte superiore a quella presente nello stomaco. Per una base debole con un pKa di 4,4 il risultato è opposto. Di conseguenza, in linea teorica, i farmaci debolmente acidi (p. es., l’aspirina) vengono assorbiti da un ambiente acido (lo stomaco) più facilmente di quanto non facciano le basi deboli (p. es., la chinidina). Tuttavia, indipendentemente dal fatto che un farmaco sia acido o basico, la maggior parte del suo assorbimento si verifica comunque nell’intestino tenue (v. Somministrazione orale, più avanti).
Leggi anche:
- Differenza tra diffusione semplice, passiva, facilitata e osmosi
- Differenza tra trasporto attivo e passivo nella membrana plasmatica
- Differenza tra endocitosi, fagocitosi, pinocitosi, esocitosi
- Differenza tra fagociti professionali e facoltativi
- Differenza tra fagocitosi ed autofagocitosi
Diffusione passiva facilitata
Per determinate molecole (p. es., il glucoso), la velocità di attraversamento delle membrane è superiore a quella prevedibile sulla base della loro bassa liposolubilità. Una delle ipotesi è che un componente di trasporto (carrier) si combini reversibilmente con la molecola del substrato sulla superficie esterna della membrana cellulare e che il complesso carrier-substrato diffonda rapidamente attraverso la membrana liberando il substrato sul versante interno. La diffusione mediata da carrier è caratterizzata dalla selettività e dalla saturabilità: il carrier trasporta soltanto i substrati con una configurazione molecolare relativamente specifica e il processo è limitato dalla disponibilità dei carrier. Questo meccanismo non richiede dispendio di energia e non consente il trasporto contro un gradiente di concentrazione.
Trasporto attivo
Questo processo è caratterizzato da selettività e saturabilità e richiede dispendio di energia da parte della cellula. I substrati possono accumularsi nel compartimento intracellulare contro un gradiente di concentrazione. Il trasporto attivo sembra essere limitato ai farmaci strutturalmente simili a sostanze endogene; questi farmaci vengono solitamente assorbiti in tratti specifici dell’intestino tenue. Processi di trasporto attivo sono stati identificati per diversi ioni, vitamine, zuccheri e aminoacidi.
Pinocitosi
E’ il meccanismo con il quale le cellule incorporano materiale liquido o particelle solide. La membrana cellulare si invagina, circonda il fluido o le particelle e quindi si fonde di nuovo formando una vescicola che in seguito si distacca e si muove verso l’interno della cellula. Anche questo meccanismo richiede dispendio di energia. La pinocitosi riveste probabilmente un ruolo marginale nel trasporto dei farmaci, se si eccettuano quelli di natura proteica.
Per approfondire:
- Assorbimento dei farmaci: somministrazione orale, parenterale e forme a rilascio controllato
- Biodisponibilità di un farmaco: cause di bassa biodisponibilità e valutazione
- Distribuzione, velocità di ingresso, equilibrio di distribuzione e legame di un farmaco
- Finestra terapeutica ed esempi di farmaci con ampi e ristretti indici terapeutici
- Eliminazione di un farmaco: metabolismo, citocromo P-450, coniugazione
- Eliminazione di un farmaco: escrezione renale e biliare
- Farmacocinetica, biodisponibilità, volume di distribuzione, emivita di un farmaco e variabilità individuale
- Farmacodinamica, interazioni farmaco-recettore e relazione dose-risposta
- Farmacogenetica: variabilità farmacocinetica e farmacodinamica
- Interazioni farmacologiche: farmacodinamiche e farmacocinetiche
- Reazioni avverse ai farmaci: effetti collaterali, tossicità, allergie, idiosincrasie
- Rapporto rischi-benefici nell’assunzione dei farmaci
- Mancata compliance del paziente: quando il paziente non prende i suoi farmaci
Leggi anche:
- Differenza tra farmacodinamica e farmacocinetica
- Il paracetamolo (Tachipirina) è un antinfiammatorio non steroideo (FANS)?
- Antinfiammatori non steroidei (FANS): significato ed elenco di farmaci
- Differenze tra Paracetamolo, Tachipirina, Ibuprofene, Aspirina, Efferalgan e Co-Efferalgan
- Comportamento dei farmaci nella barriera emato-encefalica
- Meglio Aspirina o Ibuprofene?
- A che serve la Tachipirina (paracetamolo)?
- Differenza tra farmaco originale, generico ed equivalente
- Che significa farmaco non steroideo?
- Che significa il termine “farmaco”: definizione ed etimologia
- Differenza tra farmaci di fascia A, B, C, H
- Differenza tra farmaco etico, da banco, senza obbligo di ricetta, speciale e limitativa
- Differenza tra farmaco a rilascio prolungato, ritardato, modificato, ripetuto, controllato
- Che significa farmaco di fascia C o Classe C?
- Che significa farmaco etico, esempio, classe A, è detraibile, elenco
- Farmaco “ideale”: quali sono le sue caratteristiche?
- Che significa farmaco da banco?
- Differenza tra farmaco etico e generico
- Che significa farmaco mutuabile?
- Che significa farmaco ripetibile? Caratteristiche della ricetta
- Cos’è un farmaco biologico, a che serve e come funziona?
- Che significa farmaco generico o equivalente?
- Antiossidanti: alimenti ed integratori migliori contro i radicali liberi
- Sali minerali: definizione, funzioni, alimenti, integratori [GUIDA COMPLETA]
- Differenza tra omeopatia, fitoterapia ed erboristeria
- Differenza tra Eutirox e Ibsa nella cura dell’ipotiroidismo
- Ipertensione: quali farmaci usare per abbassare la pressione arteriosa?
- Coumadin: quando si usa, dosaggio ed effetti collaterali (foglio illustrativo)
- Cos’è una sostanza stupefacente?
- Gli integratori alimentari sono doping? Elenco delle sostanze dopanti
- Differenza tra integratori alimentari e sostanze dopanti con esempi
- Differenze tra effetto collaterale, effetti indesiderati, reazione avversa, evento avverso
- Differenza tra controindicazioni ed effetti indesiderati
- Che significa “ai pasti”? Quando assumere i farmaci?
- Che significa “effetto placebo” e perché un placebo funziona?
- Floriterapia e Fiori di Bach: elenco, classificazione, funzioni
- Omeopatia: cos’è? I farmaci omeopatici funzionano realmente?
- Stitichezza o stipsi acuta e cronica: terapie farmacologiche
- Reflusso gastroesofageo: terapia farmacologica e chirurgica
- I migliori farmaci antiacidi da banco, senza ricetta medica
- Eutirox può essere usato anche per dimagrire? Quali sono i rischi?
- Eutirox: effetti collaterali e controindicazioni del farmaco
- Come fare un clistere evacuativo: procedura semplice con enteroclisma
- Fare un clistere evacuativo: procedura semplice con peretta
- Microclisma: cos’è e come si usa in adulti e neonati
- Differenze tra clistere, peretta, enteroclisma, microclisma
- Guida facile per fare una iniezione intramuscolare corretta ed indolore
- In quali parti del corpo e dove si esegue una iniezione intramuscolare?
- Com’è fatta una siringa e come si usa correttamente?
- Differenza tra dolore acuto, cronico, persistente ed episodico con esempi
- Differenza tra dolore somatico e psicosomatico
- Dolore: come si misura? La scala visiva e numerica verbale
- Dolore: quando chiamare il medico e cosa riferirgli
- Dolore: esistono esami specifici per rilevarlo?
- Meglio Aspirina o Ibuprofene?
- Supposta rettale: vantaggi e svantaggi rispetto ad altre vie di somministrazione
- Cosa succede ad una supposta dopo averla inserita? Come funziona?
- Supposte di glicerina: come usarle in bambini, adulti, gravidanza
- Come mettere facilmente una supposta a neonati, bambini, adulti
- Si possono tagliare o spezzare le supposte rettali?
- Lingua bianca, impastata, spaccata: cause e quando è pericolosa
- Vie di somministrazione di un farmaco: tipi, differenze, vantaggi e svantaggi
- A che serve la Tachipirina (paracetamolo)?
- Gravidanza e allattamento: posso assumere Tachipirina, Ibuprofene e Co-Efferalgan? Quante compresse?
- Differenze tra Efferalgan e Co-Efferalgan
- Cardioaspirin 100mg: effetti indesiderati, a cosa serve, dosaggi (foglio illustrativo)
- Che significa somministrazione di un farmaco PER OS o PO?
- Via di somministrazione orale, per os: vantaggi e svantaggi
- Tachipirina, paracetamolo, Efferalgan: posologia, controindicazioni ed effetti collaterali
- Voltaren Emulgel (diclofenac): come usarlo, gravidanza ed effetti collaterali
- Arvenum: terapia di emorroidi e fragilità capillari
- Enterogermina per gonfiore, diarrea e dolori addominali: foglietto illustrativo
- Moment (ibuprofene): posologia, effetti collaterali, gravidanza, prezzo
- Rinazina spray nasale in bambini e adulti: posologia e prezzo
- Glicerolo Carlo Erba soluzione rettale e supposte: posologia ed effetti collaterali
- Triatec (ramipril): posologia, effetti collaterali [FOGLIETTO ILLUSTRATIVO]
- Lasix (furosemide): posologia, effetti collaterali [FOGLIETTO ILLUSTRATIVO]
- Norvasc (amlodipina): posologia, effetti collaterali [FOGLIETTO ILLUSTRATIVO]
- Augmentin (amoxicillina): posologia, effetti collaterali [FOGLIETTO ILLUSTRATIVO]
- Omeprazen (omeprazolo): posologia, effetti collaterali [FOGLIETTO ILLUSTRATIVO]
- Dibase (vitamina D): posologia, effetti collaterali [FOGLIETTO ILLUSTRATIVO]
- Torvast (atorvastatina): posologia, effetti collaterali [FOGLIETTO ILLUSTRATIVO]
- Differenza tra Gabapentin e Pregabalin
- Farmaco Lyrica (Pregabalin): indicazioni ed effetti collaterali
- Lyrica (Pregabalin): è un farmaco che fa ingrassare?
Lo Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o unisciti al nostro gruppo Facebook o ancora seguici su Twitter, su Instagram o su Pinterest, grazie!
