
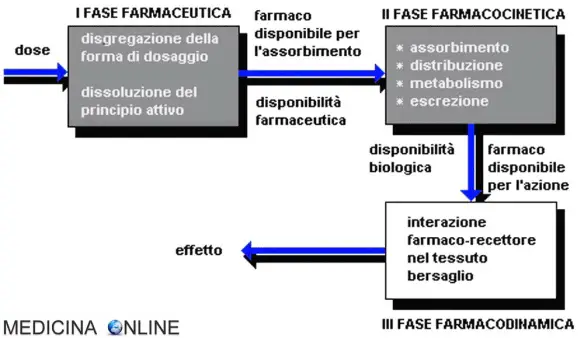
 La farmacodinamica è lo studio degli effetti biochimici e fisiologici dei farmaci e dei loro meccanismi di azione. Molti farmaci producono risposte farmacologiche interagendo (legandosi) con macromolecole specifiche, solitamente proteine complesse, sulla superficie o all’interno delle cellule. Alcune classi di farmaci reagiscono direttamente con sostanze non proteiche endogene o esogene; rientrano in questa categoria alcuni farmaci chemioterapici antitumorali che interagiscono con gli acidi nucleici, i farmaci chelanti dei metalli (p. es., l’edetato disodico di calcio, il dimercaprolo, la deferoxamina) e gli antiacidi utilizzati per neutralizzare chimicamente l’acidità gastrica.
La farmacodinamica è lo studio degli effetti biochimici e fisiologici dei farmaci e dei loro meccanismi di azione. Molti farmaci producono risposte farmacologiche interagendo (legandosi) con macromolecole specifiche, solitamente proteine complesse, sulla superficie o all’interno delle cellule. Alcune classi di farmaci reagiscono direttamente con sostanze non proteiche endogene o esogene; rientrano in questa categoria alcuni farmaci chemioterapici antitumorali che interagiscono con gli acidi nucleici, i farmaci chelanti dei metalli (p. es., l’edetato disodico di calcio, il dimercaprolo, la deferoxamina) e gli antiacidi utilizzati per neutralizzare chimicamente l’acidità gastrica.
INTERAZIONI FARMACO-RECETTORE
Praticamente nessun farmaco possiede specificità assoluta: la maggioranza dei farmaci ha invece relativa selettività; p. es., l’atropina inibisce le azioni dell’acetilcolina sulle ghiandole esocrine e sulla muscolatura liscia, ma non quelle sulla muscolatura scheletrica. L’azione di tali farmaci selettivi è dovuta al loro legame fisico-chimico con componenti cellulari denominati recettori. I recettori fisiologici sono macromolecole implicate nella trasmissione chimica dei segnali tra una cellula e l’altra e all’interno delle cellule. Una molecola che si lega a un recettore è definita ligando. Quando un ligando (ormone, neurotrasmettitore, messaggero intracellulare o farmaco esogeno) si combina con un recettore, la funzione cellulare viene modificata; ciascun ligando può interagire con più sottotipi di recettori. I recettori attivati regolano direttamente o indirettamente i processi biochimici cellulari (p. es., la conduttanza ionica, la fosforilazione proteica, la trascrizione del DNA). In molti casi, i recettori situati all’interno della membrana cellulare sono accoppiati per mezzo di proteine leganti il guanin nucleotide (proteine G) con vari sistemi effettori cui partecipano molecole intracellulari che funzionano da secondi messaggeri.
I recettori sono strutture dinamiche, influenzate sia da fattori esterni sia da meccanismi regolatori intracellulari. La up-regulation e la down-regulation dei recettori riguardano fenomeni di adattamento ai farmaci i quali possiedono importanti implicazioni cliniche (desensibilizzazione, tachifilassi, tolleranza, resistenza acquisita, ipersensibilità da sospensione).
Le specifiche regioni molecolari delle macromolecole recettoriali alle quali si lega il ligando vengono dette siti di riconoscimento. Un farmaco può interagire a livello dello stesso sito con il quale interagisce un agonista endogeno (ormone o neurotrasmettitore), oppure a livello di un sito diverso. Gli agonisti che si legano a siti adiacenti o differenti sono talvolta denominati agonisti allosterici. I farmaci vengono legati anche in modo non specifico, cioè a livello di siti molecolari non connotati come recettori (p. es., le proteine plasmatiche).
La teoria recettoriale dei farmaci, basata sulla legge dell’azione di massa, è in qualche modo paragonabile alle analisi cinetiche dell’interazione e dell’inibizione tra enzimi e substrati. Molti meccanismi biochimici dei farmaci possono essere studiati nell’ambito di questo modello di riferimento (p. es., le interazioni tra aspirina e inibitore della prostaglandina sintetasi, tra neostigmina e inibitore della colinesterasi, tra selegilina e inibitore della monoaminossidasi B). La teoria recettoriale dei farmaci include i concetti di affinità (la probabilità che un farmaco occupi un recettore in un determinato momento) e di efficacia intrinseca (attività intrinseca), che esprime le complesse associazioni tra la concentrazione del farmaco o del ligando, gli stati di attivazione dei recettori e la risposta funzionale cellulare o tissutale.
Le funzioni fisiologiche (p. es., la contrazione, la secrezione) sono regolate da meccanismi multipli mediati da recettori e di conseguenza possono essere modulate da stimoli molecolari differenti. Tra l’interazione molecolare iniziale farmaco-recettore e la risposta finale tissutale od organica vi può essere l’interposizione di diverse tappe (che coinvolgono p. es., l’accoppiamento recettoriale e l’intervento di secondi messaggeri intracellulari multipli). La densità dei recettori e l’efficienza dei meccanismi di risposta allo stimolo variano da tessuto a tessuto.
La teoria dell’occupazione precoce dei farmaci assumeva che una risposta farmacologica fosse direttamente proporzionale all’occupazione dei recettori; si riteneva che quando tutti i recettori fossero stati occupati o attivati si verificasse un effetto massimale. La teoria attuale prevede il coinvolgimento di processi cinetici (velocità di inizio/fine) relativi all’occupazione del recettore da parte del ligando, di stati di attivazione multipli dei recettori (attivo/inattivo) e della mancanza di un’evidente proporzionalità tra l’occupazione del recettore da parte del ligando e la risposta finale tissutale od organica. In questi modelli vengono prese in considerazione le variazioni nell’efficienza della trasduzione del segnale (meccanismi di amplificazione cellulare) e l’esistenza dei recettori di riserva, degli agonisti parziali e degli agonisti inversi.
I farmaci agonisti interagiscono con i recettori in modo da modificare la proporzione dei recettori attivati, modificando così l’attività cellulare. Gli agonisti convenzionali aumentano la proporzione dei recettori attivati; gli agonisti inversi la riducono. Molti ormoni e neurotrasmettitori (p. es., l’acetilcolina, l’istamina, la norepinefrina) e molti farmaci (p. es., la morfina, la fenilefrina, l’isoproterenolo) agiscono come agonisti.
Gli antagonisti interagiscono selettivamente con i recettori, ma non determinano un effetto osservabile; essi riducono l’azione di un’altra sostanza (l’agonista) a livello del sito recettoriale interessato. Gli antagonisti recettoriali sono quindi dotati di affinità ma sono privi di efficacia intrinseca.
Gli analoghi strutturali delle molecole degli agonisti possiedono frequentemente proprietà bivalenti agoniste e antagoniste; tali farmaci sono definiti agonisti parziali (a bassa efficacia). Per esempio, per i recettori b-adrenergici di alcuni tessuti, l’isoproterenolo è un agonista completo e il prenalterolo è un agonista parziale. Un farmaco che agisce come agonista parziale a livello di un tessuto può agire come agonista completo a livello di un altro tessuto.
Gli antagonisti recettoriali possono essere classificati come reversibili o irreversibili. Gli antagonisti reversibili si dissociano facilmente dal loro recettore; gli antagonisti irreversibili formano con esso un legame chimico stabile (come avviene p. es., nell’alchilazione). Gli antagonisti pseudoirreversibili si dissociano lentamente dal loro recettore. Nell’antagonismo competitivo, il legame dell’agonista e dell’antagonista è reciprocamente esclusivo, probabilmente perché entrambi gli agenti si legano allo stesso sito recettoriale. Nell’antagonismo non-competitivo, l’agonista e l’antagonista possono venire legati contemporaneamente, ma il legame dell’antagonista riduce o inibisce l’azione dell’agonista. Nell’antagonismo competitivo reversibile, l’agonista e l’antagonista formano legami di breve durata con il recettore e tra agonista, antagonista e recettore viene raggiunto uno stato di equilibrio. Tale antagonismo può essere superato aumentando la concentrazione dell’agonista; in altre parole, l’antagonismo è sormontabile. Per esempio il naloxone, un antagonista dei recettori per gli oppioidi strutturalmente simile alla morfina, dotato di scarsa o nulla attività morfino-simile, blocca gli effetti della morfina quando viene somministrato prima o dopo di essa. Tuttavia, l’antagonismo competitivo del naloxone può essere superato somministrando una maggiore quantità di morfina.
RELAZIONE DOSE-RISPOSTA
La relazione dose-risposta è la corrispondenza tra la quantità di farmaco somministrata e l’entità della risposta suscitata. La relazione dose-risposta ha implicazioni molto importanti per le decisioni terapeutiche e la farmacologia sperimentale. Il rapporto dose-risposta viene tipicamente descritto con un grafico nel quale l’effetto misurato (risposta) viene riportato sull’asse delle ordinate e la dose o una funzione della dose (p. es., il suo log10) viene riportato sull’asse delle ascisse. Poiché un effetto farmacologico è una funzione sia della dose (o concentrazione) sia del tempo, un grafico di questo tipo descrive la relazione dose-risposta in maniera indipendente dal tempo. Gli effetti misurati vengono frequentemente registrati come punti di massimo al momento dell’effetto di picco o allo stato stazionario (p. es., durante l’infusione EV continua). Gli effetti farmacologici possono essere quantificati a livello molecolare, cellulare, tissutale, organico, di apparato o dell’intero organismo.
Una curva dose-risposta ipotetica possiede caratteristiche variabili: potenza (posizione della curva lungo l’asse della dose), efficacia massima o effetto massimo (la più intensa risposta raggiungibile) e pendenza (variazione della risposta per unità di dose). Esiste anche una variazione biologica (variazione dell’intensità della risposta tra individui di controllo appartenenti alla stessa popolazione ai quali è stata somministrata la stessa dose di farmaco). Costruire curve dose-risposta per farmaci che vengono studiati in condizioni sperimentali identiche può essere di aiuto per confrontare i loro profili farmacologici.
Per approfondire:
- Assorbimento dei farmaci: somministrazione orale, parenterale e forme a rilascio controllato
- Diffusione attiva, passiva o pinocitosi: il trasporto dei farmaci attraverso le membrane cellulari
- Biodisponibilità di un farmaco: cause di bassa biodisponibilità e valutazione
- Distribuzione, velocità di ingresso, equilibrio di distribuzione e legame di un farmaco
- Finestra terapeutica ed esempi di farmaci con ampi e ristretti indici terapeutici
- Eliminazione di un farmaco: metabolismo, citocromo P-450, coniugazione
- Eliminazione di un farmaco: escrezione renale e biliare
- Farmacocinetica, biodisponibilità, volume di distribuzione, emivita di un farmaco e variabilità individuale
- Farmacogenetica: variabilità farmacocinetica e farmacodinamica
- Interazioni farmacologiche: farmacodinamiche e farmacocinetiche
- Reazioni avverse ai farmaci: effetti collaterali, tossicità, allergie, idiosincrasie
- Rapporto rischi-benefici nell’assunzione dei farmaci
- Mancata compliance del paziente: quando il paziente non prende i suoi farmaci
Leggi anche:
- Differenza tra farmacodinamica e farmacocinetica
- Il paracetamolo (Tachipirina) è un antinfiammatorio non steroideo (FANS)?
- Antinfiammatori non steroidei (FANS): significato ed elenco di farmaci
- Differenze tra Paracetamolo, Tachipirina, Ibuprofene, Aspirina, Efferalgan e Co-Efferalgan
- Comportamento dei farmaci nella barriera emato-encefalica
- Meglio Aspirina o Ibuprofene?
- A che serve la Tachipirina (paracetamolo)?
- Differenza tra farmaco originale, generico ed equivalente
- Che significa farmaco non steroideo?
- Che significa il termine “farmaco”: definizione ed etimologia
- Differenza tra farmaci di fascia A, B, C, H
- Differenza tra farmaco etico, da banco, senza obbligo di ricetta, speciale e limitativa
- Differenza tra farmaco a rilascio prolungato, ritardato, modificato, ripetuto, controllato
- Che significa farmaco di fascia C o Classe C?
- Che significa farmaco etico, esempio, classe A, è detraibile, elenco
- Farmaco “ideale”: quali sono le sue caratteristiche?
- Che significa farmaco da banco?
- Differenza tra farmaco etico e generico
- Che significa farmaco mutuabile?
- Che significa farmaco ripetibile? Caratteristiche della ricetta
- Cos’è un farmaco biologico, a che serve e come funziona?
- Che significa farmaco generico o equivalente?
- Antiossidanti: alimenti ed integratori migliori contro i radicali liberi
- Sali minerali: definizione, funzioni, alimenti, integratori [GUIDA COMPLETA]
- Differenza tra omeopatia, fitoterapia ed erboristeria
- Differenza tra Eutirox e Ibsa nella cura dell’ipotiroidismo
- Ipertensione: quali farmaci usare per abbassare la pressione arteriosa?
- Coumadin: quando si usa, dosaggio ed effetti collaterali (foglio illustrativo)
- Cos’è una sostanza stupefacente?
- Gli integratori alimentari sono doping? Elenco delle sostanze dopanti
- Differenza tra integratori alimentari e sostanze dopanti con esempi
- Differenze tra effetto collaterale, effetti indesiderati, reazione avversa, evento avverso
- Differenza tra controindicazioni ed effetti indesiderati
- Che significa “ai pasti”? Quando assumere i farmaci?
- Che significa “effetto placebo” e perché un placebo funziona?
- Floriterapia e Fiori di Bach: elenco, classificazione, funzioni
- Omeopatia: cos’è? I farmaci omeopatici funzionano realmente?
- Stitichezza o stipsi acuta e cronica: terapie farmacologiche
- Reflusso gastroesofageo: terapia farmacologica e chirurgica
- I migliori farmaci antiacidi da banco, senza ricetta medica
- Eutirox può essere usato anche per dimagrire? Quali sono i rischi?
- Eutirox: effetti collaterali e controindicazioni del farmaco
- Come fare un clistere evacuativo: procedura semplice con enteroclisma
- Fare un clistere evacuativo: procedura semplice con peretta
- Microclisma: cos’è e come si usa in adulti e neonati
- Differenze tra clistere, peretta, enteroclisma, microclisma
- Guida facile per fare una iniezione intramuscolare corretta ed indolore
- In quali parti del corpo e dove si esegue una iniezione intramuscolare?
- Com’è fatta una siringa e come si usa correttamente?
- Differenza tra dolore acuto, cronico, persistente ed episodico con esempi
- Differenza tra dolore somatico e psicosomatico
- Dolore: come si misura? La scala visiva e numerica verbale
- Dolore: quando chiamare il medico e cosa riferirgli
- Dolore: esistono esami specifici per rilevarlo?
- Meglio Aspirina o Ibuprofene?
- Supposta rettale: vantaggi e svantaggi rispetto ad altre vie di somministrazione
- Cosa succede ad una supposta dopo averla inserita? Come funziona?
- Supposte di glicerina: come usarle in bambini, adulti, gravidanza
- Come mettere facilmente una supposta a neonati, bambini, adulti
- Si possono tagliare o spezzare le supposte rettali?
- Lingua bianca, impastata, spaccata: cause e quando è pericolosa
- Vie di somministrazione di un farmaco: tipi, differenze, vantaggi e svantaggi
- A che serve la Tachipirina (paracetamolo)?
- Gravidanza e allattamento: posso assumere Tachipirina, Ibuprofene e Co-Efferalgan? Quante compresse?
- Differenze tra Efferalgan e Co-Efferalgan
- Cardioaspirin 100mg: effetti indesiderati, a cosa serve, dosaggi (foglio illustrativo)
- Che significa somministrazione di un farmaco PER OS o PO?
- Via di somministrazione orale, per os: vantaggi e svantaggi
- Tachipirina, paracetamolo, Efferalgan: posologia, controindicazioni ed effetti collaterali
- Voltaren Emulgel (diclofenac): come usarlo, gravidanza ed effetti collaterali
- Arvenum: terapia di emorroidi e fragilità capillari
- Enterogermina per gonfiore, diarrea e dolori addominali: foglietto illustrativo
- Moment (ibuprofene): posologia, effetti collaterali, gravidanza, prezzo
- Rinazina spray nasale in bambini e adulti: posologia e prezzo
- Glicerolo Carlo Erba soluzione rettale e supposte: posologia ed effetti collaterali
- Triatec (ramipril): posologia, effetti collaterali [FOGLIETTO ILLUSTRATIVO]
- Lasix (furosemide): posologia, effetti collaterali [FOGLIETTO ILLUSTRATIVO]
- Norvasc (amlodipina): posologia, effetti collaterali [FOGLIETTO ILLUSTRATIVO]
- Augmentin (amoxicillina): posologia, effetti collaterali [FOGLIETTO ILLUSTRATIVO]
- Omeprazen (omeprazolo): posologia, effetti collaterali [FOGLIETTO ILLUSTRATIVO]
- Dibase (vitamina D): posologia, effetti collaterali [FOGLIETTO ILLUSTRATIVO]
- Torvast (atorvastatina): posologia, effetti collaterali [FOGLIETTO ILLUSTRATIVO]
- Differenza tra Gabapentin e Pregabalin
- Farmaco Lyrica (Pregabalin): indicazioni ed effetti collaterali
- Lyrica (Pregabalin): è un farmaco che fa ingrassare?
Lo Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o unisciti al nostro gruppo Facebook o ancora seguici su Twitter, su Instagram o su Pinterest, grazie!
