
 La farmacocinetica è lo studio dell’andamento temporale delle modificazioni cui un farmaco e i suoi metaboliti vanno incontro all’interno dell’organismo, dopo l’assunzione attraverso qualunque via di somministrazione.
La farmacocinetica è lo studio dell’andamento temporale delle modificazioni cui un farmaco e i suoi metaboliti vanno incontro all’interno dell’organismo, dopo l’assunzione attraverso qualunque via di somministrazione.
Perché si abbia una risposta appropriata a un farmaco, è necessario che esso sia presente in concentrazione adeguata a livello del sito di azione. Il regime di dosaggio richiesto per raggiungere e mantenere tale concentrazione dipende dalla farmacocinetica. La concentrazione appropriata e il regime posologico dipendono dalle condizioni cliniche del paziente, dalla gravità della patologia, dalla presenza di malattie concomitanti, dall’uso di altri farmaci e da altri fattori ancora.
A causa delle differenze individuali, la somministrazione dei farmaci deve essere basata sulle esigenze di ogni singolo paziente, il che viene da sempre ottenuto modificando empiricamente il dosaggio finché non si raggiunge l’obiettivo terapeutico desiderato. Questo approccio è spesso inadeguato, perché la risposta ottimale può essere ritardata o possono verificarsi reazioni tossiche gravi. In alternativa, un farmaco può essere somministrato sulla base dell’assorbimento e della disposizione (distribuzione ed eliminazione) che si prevede esso abbia in un paziente, e la posologia può essere regolata controllando la concentrazione plasmatica del farmaco e i suoi effetti farmacologici. Questo approccio richiede la conoscenza della farmacocinetica del composto in funzione dell’età e del peso corporeo del paziente, oltre che delle conseguenze farmacocinetiche delle eventuali malattie concomitanti (p. es., malattie renali, epatiche o cardiovascolari o una combinazione di più patologie).
La biodisponibilità esprime l’entità dell’assorbimento dei farmaci nella circolazione sistemica. La costante della velocità di assorbimento esprime la velocità con cui avviene l’assorbimento. Questi parametri influenzano la concentrazione massima (di picco), il tempo necessario per raggiungere la concentrazione massima (tempo di picco) e l’area al di sotto della curva concentrazione-tempo (AUC) dopo una dose orale singola. Durante la terapia farmacologica a lungo termine, la misura più importante è l’entità dell’assorbimento, perché da essa dipende la concentrazione media; il grado di fluttuazione della concentrazione è legato alla costante della velocità di assorbimento.
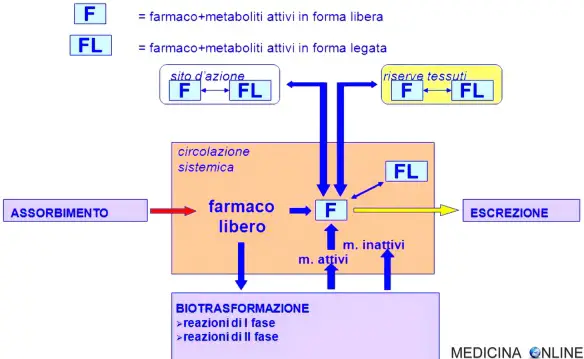
Il volume apparente di distribuzione è la quantità di liquido che sarebbe necessaria per contenere il farmaco presente nell’organismo alla stessa concentrazione alla quale esso si trova nel sangue o nel plasma. Esso può essere utilizzato per calcolare la dose necessaria per ottenere una determinata concentrazione, come pure la concentrazione attesa dopo la somministrazione di una determinata dose. La concentrazione del farmaco non legato è strettamente correlata agli effetti farmacologici, quindi la frazione libera è una misura utile, particolarmente quando il legame alle proteine plasmatiche è alterato, p. es., dall’ipoalbuminemia, da malattie renali o epatiche oppure dalla presenza di interazioni competitive. Il volume apparente di distribuzione e la frazione libera plasmatica sono i parametri più diffusamente utilizzati per la valutazione della distribuzione dei farmaci.
La velocità di eliminazione di un farmaco dall’organismo varia parallelamente alla concentrazione plasmatica. Il parametro che lega la velocità di eliminazione e la concentrazione plasmatica è la clearance totale, che equivale alla somma della clearance renale e di quella extrarenale (metabolica).
La frazione escreta immodificata è utile per la valutazione degli effetti potenziali delle patologie renali ed epatiche sull’eliminazione dei farmaci. Una frazione bassa indica che il probabile meccanismo di eliminazione è il metabolismo epatico e che una patologia epatica può quindi alterare l’eliminazione del farmaco. Le patologie renali provocano effetti più consistenti sulla cinetica dei farmaci che possiedono un’alta frazione escreta immodificata.
La velocità di estrazione di un farmaco dal sangue da parte di un organo emuntore, come il fegato, non può essere superiore alla velocità di cessione del farmaco all’organo stesso. Di conseguenza, la clearance ha un limite superiore, dipendente dalla cessione del farmaco e quindi dal flusso ematico all’organo in questione. Inoltre, quando l’organo preposto all’eliminazione è il fegato o la parete intestinale e il farmaco viene somministrato per via orale, una parte della dose può essere metabolizzata durante il passaggio attraverso i tessuti verso la circolazione sistemica; questo processo è chiamato effetto di primo passaggio. Pertanto, se l’estrazione (clearance) di un farmaco è elevata nel fegato o nella parete intestinale, la sua biodisponibilità per via orale è bassa, il che talvolta preclude l’impiego della somministrazione orale o richiede una dose orale molto più elevata rispetto a una dose parenterale equivalente. Tra i farmaci con notevole metabolismo di primo passaggio vi sono l’alprenololo, l’idralazina, l’isoproterenolo, la lidocaina, la meperidina, la morfina, la nifedipina, la nitroglicerina, il propranololo, il testosterone e il verapamil.
La costante della velocità di eliminazione è una funzione del modo in cui un farmaco viene estratto dal sangue per opera degli organi emuntori e del modo in cui il farmaco si distribuisce nell’organismo.
L’emivita (di eliminazione) è il tempo necessario perché la concentrazione plasmatica di un farmaco o la quantità di farmaco presente nell’organismo si riduca del 50%. Per la maggior parte dei farmaci, l’emivita rimane costante indipendentemente dalla quantità di farmaco presente nell’organismo. Le eccezioni comprendono la fenitoina, la teofillina e l’eparina.
Il tempo medio di permanenza (Mean Residence Time, MRT), un’altra misura dell’eliminazione dei farmaci, è il tempo medio per il quale la molecola di un farmaco permane nell’organismo dopo la sua iniezione EV rapida. Analogamente alla clearance, il suo valore è indipendente dalla dose.
VARIABILITA’ DEI VALORI DEI PARAMETRI
Al momento di adattare la somministrazione di un farmaco alle esigenze di un determinato paziente, devono essere tenuti in considerazione molti fattori in grado di modificare i parametri farmacocinetici. Tuttavia, anche con l’adattamento della posologia, di solito rimane comunque un discreto grado di variabilità; di conseguenza la risposta ai farmaci e, in alcuni casi, la loro concentrazione plasmatica, devono essere tenute sotto controllo con grande attenzione.
Età e peso: per alcuni farmaci, gli effetti dell’età e del peso sulla farmacocinetica sono ben documentati. Per gli individui di età compresa fra i 6 mesi e i 20 anni, la funzione renale appare ben correlata con l’ASC. Pertanto, per i farmaci eliminati prevalentemente per via renale in forma immodificata, la clearance nei bambini si modifica con l’età parallelamente al variare dell’ASC. Negli individui di età > 20 anni, la funzionalità renale diminuisce circa dell’1% ogni anno. Quindi, il dosaggio di questi farmaci può essere modificato in base all’età. L’ASC nei bambini è correlata anche alla clearance metabolica, sebbene le eccezioni siano frequenti. Nei neonati e nei lattanti, la funzione renale e quella epatica non sono ancora pienamente sviluppate e le generalizzazioni, al di fuori dell’evenienza di una modificazione repentina, sono meno accurate.
Compromissione della funzionalità renale: la clearance renale della maggior parte dei farmaci sembra variare in funzione diretta della clearance della creatinina, indipendentemente dal tipo di patologia renale presente. La modificazione della clearance totale dipende dal contributo dei reni all’eliminazione totale del farmaco. Di conseguenza, la clearance totale dovrebbe essere proporzionale alla funzionalità renale (clearance della creatinina) per i farmaci che vengono escreti immodificati e non dovrebbe risultare alterata per i farmaci che vengono eliminati per metabolizzazione. L’insufficienza renale può alterare il volume apparente di distribuzione, che nel caso della digossina diminuisce a causa della diminuzione del legame tissutale e nel caso della fenitoina, dell’acido salicilico e di molti altri farmaci aumenta a causa della diminuzione del legame alle proteine plasmatiche.
Leggi anche:
- Velocità di filtrazione glomerulare: valori normali, bassi e insufficienza renale
- Filtrazione glomerulare, riassorbimento e secrezione
- Glomerulo renale: schema, funzione e flusso ematico renale
- Insufficienza renale acuta: sintomi, terapia, linea guida, morte
- Insufficienza renale cronica: stadi, dieta, sintomi, diagnosi e terapia
- Esami per valutare funzionalità renale ed insufficienza renale
Stress fisiologico: la concentrazione della proteina di fase acuta a1-glicoproteina acida aumenta durante lo stress fisiologico (p. es., IMA, interventi chirurgici, colite ulcerosa, morbo di Crohn). Conseguentemente, aumenta il legame di diversi farmaci (p. es., il propranololo, la chinidina, la disopiramide) a questa proteina, e il loro volume apparente di distribuzione diminuisce di pari passo.
Malattie epatiche: una disfunzione epatica può alterare la clearance metabolica, ma finora non è stato possibile individuare fattori ben correlati o predittivi di queste modificazioni. La cirrosi epatica può ridurre criticamente il metabolismo dei farmaci e spesso provoca la riduzione del legame alle proteine plasmatiche a causa della diminuzione della albumina nel plasma. L’epatite acuta, caratterizzata dall’innalzamento degli enzimi sierici, solitamente non modifica il metabolismo dei farmaci.
Altre patologie: lo scompenso cardiaco, la polmonite, l’ipertiroidismo e molte altre condizioni patologiche possono modificare la cinetica dei farmaci.
Interazioni farmacologiche: i valori dei parametri farmacocinetici e, di conseguenza, le risposte ai farmaci possono essere influenzati dalle interazioni farmacologiche. La maggior parte delle interazioni è graduata e la loro entità dipende dalle concentrazioni di entrambi i farmaci. Pertanto, stabilire e adattare il dosaggio dei farmaci è una operazione complessa.
Dosaggio: in alcune circostanze, le modificazioni della dose, della frequenza di somministrazione o della durata della terapia alterano la cinetica di un farmaco. Per esempio, all’aumentare della dose, la biodisponibilità della griseofulvina diminuisce a causa della bassa solubilità del farmaco nei fluidi del tratto GI superiore. Per la fenitoina, la concentrazione plasmatica allo stato stazionario aumenta in maniera sproporzionata quando viene aumentata la velocità di somministrazione, dal momento che l’enzima deputato al suo metabolismo ha una limitata capacità di eliminazione del farmaco e che la velocità di somministrazione abituale si avvicina alla velocità massima di metabolizzazione. La concentrazione plasmatica della carbamazepina diminuisce durante la somministrazione prolungata, perché la carbamazepina è un induttore del suo stesso metabolismo. Altre cause di modificazioni farmacocinetiche dipendenti dal dosaggio sono la saturabilità del legame alle proteine plasmatiche e ai tessuti (p. es., per il fenilbutazone), la saturabilità della secrezione a livello renale (p. es., per la penicillina ad alte dosi) e la saturabilità del metabolismo di primo passaggio attraverso il fegato (p. es. per il propranololo).
Per approfondire:
- Assorbimento dei farmaci: somministrazione orale, parenterale e forme a rilascio controllato
- Diffusione attiva, passiva o pinocitosi: il trasporto dei farmaci attraverso le membrane cellulari
- Biodisponibilità di un farmaco: cause di bassa biodisponibilità e valutazione
- Distribuzione, velocità di ingresso, equilibrio di distribuzione e legame di un farmaco
- Finestra terapeutica ed esempi di farmaci con ampi e ristretti indici terapeutici
- Eliminazione di un farmaco: metabolismo, citocromo P-450, coniugazione
- Eliminazione di un farmaco: escrezione renale e biliare
- Farmacodinamica, interazioni farmaco-recettore e relazione dose-risposta
- Farmacogenetica: variabilità farmacocinetica e farmacodinamica
- Interazioni farmacologiche: farmacodinamiche e farmacocinetiche
- Reazioni avverse ai farmaci: effetti collaterali, tossicità, allergie, idiosincrasie
- Rapporto rischi-benefici nell’assunzione dei farmaci
- Mancata compliance del paziente: quando il paziente non prende i suoi farmaci
Leggi anche:
- Differenza tra farmacodinamica e farmacocinetica
- Il paracetamolo (Tachipirina) è un antinfiammatorio non steroideo (FANS)?
- Antinfiammatori non steroidei (FANS): significato ed elenco di farmaci
- Differenze tra Paracetamolo, Tachipirina, Ibuprofene, Aspirina, Efferalgan e Co-Efferalgan
- Comportamento dei farmaci nella barriera emato-encefalica
- Meglio Aspirina o Ibuprofene?
- A che serve la Tachipirina (paracetamolo)?
- Differenza tra farmaco originale, generico ed equivalente
- Che significa farmaco non steroideo?
- Che significa il termine “farmaco”: definizione ed etimologia
- Differenza tra farmaci di fascia A, B, C, H
- Differenza tra farmaco etico, da banco, senza obbligo di ricetta, speciale e limitativa
- Differenza tra farmaco a rilascio prolungato, ritardato, modificato, ripetuto, controllato
- Che significa farmaco di fascia C o Classe C?
- Che significa farmaco etico, esempio, classe A, è detraibile, elenco
- Farmaco “ideale”: quali sono le sue caratteristiche?
- Che significa farmaco da banco?
- Differenza tra farmaco etico e generico
- Che significa farmaco mutuabile?
- Che significa farmaco ripetibile? Caratteristiche della ricetta
- Cos’è un farmaco biologico, a che serve e come funziona?
- Che significa farmaco generico o equivalente?
- Antiossidanti: alimenti ed integratori migliori contro i radicali liberi
- Sali minerali: definizione, funzioni, alimenti, integratori [GUIDA COMPLETA]
- Differenza tra omeopatia, fitoterapia ed erboristeria
- Differenza tra Eutirox e Ibsa nella cura dell’ipotiroidismo
- Ipertensione: quali farmaci usare per abbassare la pressione arteriosa?
- Coumadin: quando si usa, dosaggio ed effetti collaterali (foglio illustrativo)
- Cos’è una sostanza stupefacente?
- Gli integratori alimentari sono doping? Elenco delle sostanze dopanti
- Differenza tra integratori alimentari e sostanze dopanti con esempi
- Differenze tra effetto collaterale, effetti indesiderati, reazione avversa, evento avverso
- Differenza tra controindicazioni ed effetti indesiderati
- Che significa “ai pasti”? Quando assumere i farmaci?
- Che significa “effetto placebo” e perché un placebo funziona?
- Floriterapia e Fiori di Bach: elenco, classificazione, funzioni
- Omeopatia: cos’è? I farmaci omeopatici funzionano realmente?
- Stitichezza o stipsi acuta e cronica: terapie farmacologiche
- Reflusso gastroesofageo: terapia farmacologica e chirurgica
- I migliori farmaci antiacidi da banco, senza ricetta medica
- Eutirox può essere usato anche per dimagrire? Quali sono i rischi?
- Eutirox: effetti collaterali e controindicazioni del farmaco
- Come fare un clistere evacuativo: procedura semplice con enteroclisma
- Fare un clistere evacuativo: procedura semplice con peretta
- Microclisma: cos’è e come si usa in adulti e neonati
- Differenze tra clistere, peretta, enteroclisma, microclisma
- Guida facile per fare una iniezione intramuscolare corretta ed indolore
- In quali parti del corpo e dove si esegue una iniezione intramuscolare?
- Com’è fatta una siringa e come si usa correttamente?
- Differenza tra dolore acuto, cronico, persistente ed episodico con esempi
- Differenza tra dolore somatico e psicosomatico
- Dolore: come si misura? La scala visiva e numerica verbale
- Dolore: quando chiamare il medico e cosa riferirgli
- Dolore: esistono esami specifici per rilevarlo?
- Meglio Aspirina o Ibuprofene?
- Supposta rettale: vantaggi e svantaggi rispetto ad altre vie di somministrazione
- Cosa succede ad una supposta dopo averla inserita? Come funziona?
- Supposte di glicerina: come usarle in bambini, adulti, gravidanza
- Come mettere facilmente una supposta a neonati, bambini, adulti
- Si possono tagliare o spezzare le supposte rettali?
- Lingua bianca, impastata, spaccata: cause e quando è pericolosa
- Vie di somministrazione di un farmaco: tipi, differenze, vantaggi e svantaggi
- A che serve la Tachipirina (paracetamolo)?
- Gravidanza e allattamento: posso assumere Tachipirina, Ibuprofene e Co-Efferalgan? Quante compresse?
- Differenze tra Efferalgan e Co-Efferalgan
- Cardioaspirin 100mg: effetti indesiderati, a cosa serve, dosaggi (foglio illustrativo)
- Che significa somministrazione di un farmaco PER OS o PO?
- Via di somministrazione orale, per os: vantaggi e svantaggi
- Tachipirina, paracetamolo, Efferalgan: posologia, controindicazioni ed effetti collaterali
- Voltaren Emulgel (diclofenac): come usarlo, gravidanza ed effetti collaterali
- Arvenum: terapia di emorroidi e fragilità capillari
- Enterogermina per gonfiore, diarrea e dolori addominali: foglietto illustrativo
- Moment (ibuprofene): posologia, effetti collaterali, gravidanza, prezzo
- Rinazina spray nasale in bambini e adulti: posologia e prezzo
- Glicerolo Carlo Erba soluzione rettale e supposte: posologia ed effetti collaterali
- Triatec (ramipril): posologia, effetti collaterali [FOGLIETTO ILLUSTRATIVO]
- Lasix (furosemide): posologia, effetti collaterali [FOGLIETTO ILLUSTRATIVO]
- Norvasc (amlodipina): posologia, effetti collaterali [FOGLIETTO ILLUSTRATIVO]
- Augmentin (amoxicillina): posologia, effetti collaterali [FOGLIETTO ILLUSTRATIVO]
- Omeprazen (omeprazolo): posologia, effetti collaterali [FOGLIETTO ILLUSTRATIVO]
- Dibase (vitamina D): posologia, effetti collaterali [FOGLIETTO ILLUSTRATIVO]
- Torvast (atorvastatina): posologia, effetti collaterali [FOGLIETTO ILLUSTRATIVO]
- Differenza tra Gabapentin e Pregabalin
- Farmaco Lyrica (Pregabalin): indicazioni ed effetti collaterali
- Lyrica (Pregabalin): è un farmaco che fa ingrassare?
Lo Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o unisciti al nostro gruppo Facebook o ancora seguici su Twitter, su Instagram o su Pinterest, grazie!
