
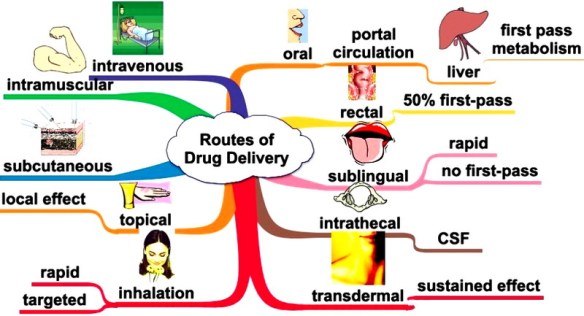
 L’assorbimento dei farmaci è determinato dalle loro proprietà fisico-chimiche, dalle loro formulazioni e dalle vie di somministrazione. I prodotti farmaceutici, cioè le effettive preparazioni (p. es., compresse, capsule, soluzioni) costituite dal farmaco e dagli eccipienti, sono formulate per essere somministrate per varie vie, tra le quali l’orale, la buccale, la sub-linguale, la rettale, la parenterale, la topica e l’inalatoria.
L’assorbimento dei farmaci è determinato dalle loro proprietà fisico-chimiche, dalle loro formulazioni e dalle vie di somministrazione. I prodotti farmaceutici, cioè le effettive preparazioni (p. es., compresse, capsule, soluzioni) costituite dal farmaco e dagli eccipienti, sono formulate per essere somministrate per varie vie, tra le quali l’orale, la buccale, la sub-linguale, la rettale, la parenterale, la topica e l’inalatoria.
Leggi anche: Vie di somministrazione di un farmaco: tipi, differenze, vantaggi e svantaggi
Somministrazione orale
Nel caso della somministrazione orale, che è la via di somministrazione più comune, l’assorbimento si riferisce al trasporto dei farmaci attraverso le membrane delle cellule epiteliali dell’apparato GI. L’assorbimento dopo la somministrazione orale è reso incostante da differenze a carico del pH intraluminale lungo il tratto GI, dell’area della superficie di assorbimento per unità di volume luminale e della perfusione ematica, oltre che dalla presenza di bile e muco e dalla natura delle membrane epiteliali. Gli acidi vengono assorbiti più rapidamente nell’intestino che nello stomaco, contraddicendo in apparenza l’ipotesi che un farmaco non ionizzato attraversa le membrane con maggior facilità. In realtà, l’apparente contraddizione è spiegata dalla più ampia superficie di assorbimento e dalla maggiore permeabilità delle membrane dell’intestino tenue.
La mucosa orale possiede un epitelio sottile e una ricca vascolarizzazione che favoriscono l’assorbimento, ma il contatto è solitamente troppo breve, anche per i farmaci in soluzione, perché abbia luogo un assorbimento apprezzabile. Un farmaco posto tra le gengive e la guancia (somministrazione buccale) o sotto la lingua (somministrazione sublinguale) viene trattenuto in situ più a lungo, consentendo un assorbimento più completo.
Lo stomaco ha una superficie epiteliale relativamente estesa, ma poiché possiede uno strato mucoso piuttosto spesso e il tempo in cui il farmaco vi staziona è di solito relativamente breve, l’assorbimento è limitato. Praticamente tutti i farmaci vengono assorbiti più velocemente dall’intestino tenue che dallo stomaco. Di conseguenza, lo svuotamento gastrico è il passaggio limitante la velocità di assorbimento. Il cibo, specialmente gli alimenti grassi, rallenta lo svuotamento gastrico (e la velocità di assorbimento dei farmaci), spiegando perché alcuni farmaci debbano essere assunti a stomaco vuoto quando si desidera un rapido inizio d’azione. Il cibo può aumentare l’entità dell’assorbimento dei farmaci scarsamente solubili (p. es., la griseofulvina), può ridurre quella dei farmaci che vengono degradati nello stomaco (p. es., la penicillina G), oppure avere effetti minimi o nulli. I farmaci che influenzano lo svuotamento gastrico (p. es., i parasimpaticolitici) modificano la velocità di assorbimento di altri farmaci.
Fra tutti i segmenti dell’apparato GI, l’intestino tenue possiede la più ampia superficie per l’assorbimento dei farmaci. Il pH intraluminale varia da 4 a 5 nel duodeno, ma diviene via via progressivamente più alcalino, avvicinandosi a 8 nell’ileo distale. La microflora GI può inattivare taluni farmaci, riducendone l’assorbimento. La riduzione del flusso ematico (p. es., nello shock) può diminuire il gradiente di concentrazione tra i due versanti della mucosa intestinale e ridurre l’assorbimento che avviene per diffusione passiva. (Anche la diminuzione del flusso ematico periferico altera la distribuzione e il metabolismo dei farmaci.)
Il tempo di transito intestinale può influenzare l’assorbimento, particolarmente dei farmaci che vengono assorbiti mediante trasporto attivo (p. es., le vitamine del gruppo B), di quelli che si disciolgono lentamente (p. es., la griseofulvina) o di quelli che sono troppo polari (cioè scarsamente liposolubili) per attraversare facilmente le membrane (p. es., molti antibiotici). Per tali farmaci, il transito può risultare troppo rapido perché l’assorbimento sia completo.
L’assorbimento delle preparazioni a rilascio controllato può avvenire principalmente nell’intestino crasso, particolarmente quando il rilascio del farmaco si protrae per più di 6 h, il tempo necessario perché il contenuto intestinale giunga nel colon.
Assorbimento dei farmaci in soluzione: un farmaco somministrato per via orale in soluzione viene a contatto con numerose secrezioni GI e, per essere assorbito, deve superare indenne l’esposizione a bassi valori di pH e a enzimi potenzialmente degradanti. Di solito, anche se un farmaco è stabile nell’ambiente intestinale, ben poco di esso rimane nel lume fino a giungere nell’intestino crasso. I farmaci poco lipofilici (cioè con scarsa capacità di attraversare le membrane), come gli aminoglicosidi, quando si trovano in soluzione vengono assorbiti lentamente nello stomaco e nell’intestino tenue; per tali farmaci, l’assorbimento a livello dell’intestino crasso è prevedibilmente ancora più lento, perché l’area della superficie di assorbimento è minore. Di conseguenza, questi farmaci non sono buoni candidati per le preparazioni a rilascio controllato.
Assorbimento dei farmaci in forma solida: la maggior parte dei farmaci viene somministrata per via orale sotto forma di compresse o capsule, principalmente per ragioni di praticità, di economia, di stabilità e di accettazione da parte del paziente. Questi prodotti devono disgregarsi e disciogliersi prima che possa avvenirne l’assorbimento. La disgregazione aumenta notevolmente la quantità di molecole di farmaco che vengono a contatto con i succhi GI, favorendo in questo modo la dissoluzione e l’assorbimento del farmaco stesso. Agenti disgreganti e altri eccipienti (p. es., diluenti, lubrificanti, surfattanti, leganti, disperdenti) vengono spesso aggiunti al farmaco durante la fabbricazione per facilitare questi processi. I surfattanti aumentano la velocità di dissoluzione incrementando la permeabilità all’acqua, la solubilità e la capacità di dispersione del farmaco. La disgregazione delle preparazioni solide può essere ritardata dall’applicazione di una pressione eccessiva durante il confezionamento delle compresse oppure da speciali rivestimenti applicati per proteggere le compresse dai processi digestivi intestinali. I lubrificanti idrofobi (p. es., lo stearato di magnesio) possono legarsi al farmaco attivo e ridurre la sua biodisponibilità.
La velocità di dissoluzione determina la maggiore o minore disponibilità del farmaco per l’assorbimento. Nel caso in cui la dissoluzione sia più lenta dell’assorbimento, essa diventa la tappa limitante la velocità del processo. L’assorbimento complessivo può essere regolato tramite modificazioni della formulazione del farmaco. Per esempio, la riduzione delle dimensioni delle particelle aumenta la superficie di contatto della sostanza, aumentando in questo modo la velocità e il grado dell’assorbimento GI di un farmaco il cui assorbimento è normalmente limitato da una lenta dissoluzione. La velocità di dissoluzione è diversa a seconda che il farmaco sia in forma salina, cristallina o idrata. I sali di Na degli acidi deboli (p. es., barbiturici, salicilati) si dissolvono più rapidamente dei loro corrispondenti acidi liberi, indipendentemente dal pH del mezzo. Alcuni farmaci sono polimorfici, esistendo in forme amorfe o in forme cristalline di vario tipo. Il cloramfenicolo palmitato esiste in due forme, ma soltanto una di esse si dissolve e viene assorbita in grado sufficiente per essere clinicamente utile. Un idrato si forma quando una o più molecole di acqua si combinano con una molecola di un farmaco in forma cristallina. La solubilità di tale solvato può essere molto differente da quella della forma non solvata; p. es., l’ampicillina anidra ha una velocità di dissoluzione e di assorbimento più elevata rispetto alla sua corrispondente forma triidrata.
Somministrazione parenterale
L’introduzione diretta di un farmaco nel torrente circolatorio (solitamente EV) assicura l’arrivo nella circolazione sistemica dell’intera dose somministrata. Il trasferimento di tutta la dose non è però garantito se una via di somministrazione richiede il passaggio attraverso una o più membrane biologiche per raggiungere la circolazione sistemica (iniezione IM o SC). Per i farmaci proteici con una massa molecolare > 20000 g/mol, il passaggio attraverso le membrane capillari è così lento, che dopo una somministrazione IM o SC la maggior parte dell’assorbimento avviene per sottrazione attraverso il sistema linfatico. In questi casi, la velocità di trasporto nella circolazione sistemica è bassa e spesso incompleta a causa del metabolismo di primo passaggio per opera degli enzimi proteolitici presenti nei vasi linfatici.
Poiché i capillari tendono a essere altamente permeabili, la perfusione (flusso ematico/grammo di tessuto) influenza notevolmente la velocità di assorbimento delle molecole di piccole dimensioni. Quindi, la sede di iniezione può avere un effetto considerevole sulla velocità di assorbimento di un farmaco; p. es., la velocità di assorbimento del diazepam iniettato IM in una sede con scarso flusso ematico può essere molto inferiore a quella che si osserva dopo somministrazione orale.
L’assorbimento può essere ritardato o irregolare quando vengono iniettati IM i sali di acidi e di basi scarsamente solubili. La forma parenterale della fenitoina è una soluzione al 40% del suo sale sodico in glicole propilenico, con un pH di circa 12. Quando la soluzione viene iniettata IM, il glicole propilenico viene assorbito e i liquidi tissutali, agendo come un tampone, riducono il pH, provocando uno spostamento dell’equilibrio tra la forma ionizzata e la forma acida libera del farmaco. Quindi l’acido libero, scarsamente solubile, precipita. Il risultato è che la dissoluzione e l’assorbimento impiegano da 1 a 2 settimane per completarsi.
Forme a rilascio controllato
Le preparazioni a rilascio controllato hanno lo scopo di ridurre la frequenza delle somministrazioni e di diminuire le fluttuazioni della concentrazione plasmatica dei farmaci, in modo da garantire un effetto terapeutico più uniforme. Una somministrazione meno frequente è più pratica e può migliorare la compliance del paziente. Queste preparazioni trovano un impiego ideale per i farmaci che altrimenti richiederebbero somministrazioni frequenti a causa della brevità della loro emivita di eliminazione e della durata del loro effetto.
Le forme a rilascio controllato destinate alla somministrazione orale sono spesso formulate in modo da mantenere le concentrazioni terapeutiche del farmaco per un periodo pari o superiore a 12 h. La velocità di assorbimento può essere controllata rivestendo le particelle del farmaco con sostanze cerose o con altri materiali non idrosolubili, includendo il farmaco in una matrice dalla quale viene liberato lentamente durante il transito attraverso il tratto GI, oppure complessando il farmaco con resine a scambio ionico.
Le preparazioni a rilascio controllato per uso transdermico hanno lo scopo di garantire il rilascio del farmaco per periodi prolungati; p. es., la diffusione della clonidina attraverso una membrana assicura la cessione controllata del farmaco per una settimana, e un polimero impregnato di nitroglicerina adsorbito su un cerotto adesivo consente la cessione controllata del farmaco per 24 h. I farmaci a rilascio transdermico devono possedere appropriate capacità di penetrazione cutanea e notevole potenza, perché il tasso di penetrazione e l’area di applicazione sono limitati.
Molte preparazioni parenterali non endovenose sono formulate in modo da mantenere elevati nel tempo i livelli ematici. Per gli antibiotici, i sali relativamente insolubili (p. es., la penicillina G benzatina) iniettati IM garantiscono il mantenimento di concentrazioni terapeutiche per periodi prolungati. Per altri farmaci, vengono formulate sospensioni o soluzioni in veicoli non acquosi (p. es., le iniezioni di insulina in sospensioni cristalline). L’insulina amorfa, dotata di un’elevata superficie di contatto per la dissoluzione, ha un rapido inizio e una breve durata di azione.
Per approfondire:
- Diffusione attiva, passiva o pinocitosi: il trasporto dei farmaci attraverso le membrane cellulari
- Biodisponibilità di un farmaco: cause di bassa biodisponibilità e valutazione
- Distribuzione, velocità di ingresso, equilibrio di distribuzione e legame di un farmaco
- Finestra terapeutica ed esempi di farmaci con ampi e ristretti indici terapeutici
- Eliminazione di un farmaco: metabolismo, citocromo P-450, coniugazione
- Eliminazione di un farmaco: escrezione renale e biliare
- Farmacocinetica, biodisponibilità, volume di distribuzione, emivita di un farmaco e variabilità individuale
- Farmacodinamica, interazioni farmaco-recettore e relazione dose-risposta
- Farmacogenetica: variabilità farmacocinetica e farmacodinamica
- Interazioni farmacologiche: farmacodinamiche e farmacocinetiche
- Reazioni avverse ai farmaci: effetti collaterali, tossicità, allergie, idiosincrasie
- Rapporto rischi-benefici nell’assunzione dei farmaci
- Mancata compliance del paziente: quando il paziente non prende i suoi farmaci
Leggi anche:
- Differenza tra farmacodinamica e farmacocinetica
- Il paracetamolo (Tachipirina) è un antinfiammatorio non steroideo (FANS)?
- Antinfiammatori non steroidei (FANS): significato ed elenco di farmaci
- Differenze tra Paracetamolo, Tachipirina, Ibuprofene, Aspirina, Efferalgan e Co-Efferalgan
- Comportamento dei farmaci nella barriera emato-encefalica
- Meglio Aspirina o Ibuprofene?
- A che serve la Tachipirina (paracetamolo)?
- Differenza tra farmaco originale, generico ed equivalente
- Che significa farmaco non steroideo?
- Che significa il termine “farmaco”: definizione ed etimologia
- Differenza tra farmaci di fascia A, B, C, H
- Differenza tra farmaco etico, da banco, senza obbligo di ricetta, speciale e limitativa
- Differenza tra farmaco a rilascio prolungato, ritardato, modificato, ripetuto, controllato
- Che significa farmaco di fascia C o Classe C?
- Che significa farmaco etico, esempio, classe A, è detraibile, elenco
- Farmaco “ideale”: quali sono le sue caratteristiche?
- Che significa farmaco da banco?
- Differenza tra farmaco etico e generico
- Che significa farmaco mutuabile?
- Che significa farmaco ripetibile? Caratteristiche della ricetta
- Cos’è un farmaco biologico, a che serve e come funziona?
- Che significa farmaco generico o equivalente?
- Antiossidanti: alimenti ed integratori migliori contro i radicali liberi
- Sali minerali: definizione, funzioni, alimenti, integratori [GUIDA COMPLETA]
- Differenza tra omeopatia, fitoterapia ed erboristeria
- Differenza tra Eutirox e Ibsa nella cura dell’ipotiroidismo
- Ipertensione: quali farmaci usare per abbassare la pressione arteriosa?
- Coumadin: quando si usa, dosaggio ed effetti collaterali (foglio illustrativo)
- Cos’è una sostanza stupefacente?
- Gli integratori alimentari sono doping? Elenco delle sostanze dopanti
- Differenza tra integratori alimentari e sostanze dopanti con esempi
- Differenze tra effetto collaterale, effetti indesiderati, reazione avversa, evento avverso
- Differenza tra controindicazioni ed effetti indesiderati
- Che significa “ai pasti”? Quando assumere i farmaci?
- Che significa “effetto placebo” e perché un placebo funziona?
- Floriterapia e Fiori di Bach: elenco, classificazione, funzioni
- Omeopatia: cos’è? I farmaci omeopatici funzionano realmente?
- Stitichezza o stipsi acuta e cronica: terapie farmacologiche
- Reflusso gastroesofageo: terapia farmacologica e chirurgica
- I migliori farmaci antiacidi da banco, senza ricetta medica
- Eutirox può essere usato anche per dimagrire? Quali sono i rischi?
- Eutirox: effetti collaterali e controindicazioni del farmaco
- Come fare un clistere evacuativo: procedura semplice con enteroclisma
- Fare un clistere evacuativo: procedura semplice con peretta
- Microclisma: cos’è e come si usa in adulti e neonati
- Differenze tra clistere, peretta, enteroclisma, microclisma
- Guida facile per fare una iniezione intramuscolare corretta ed indolore
- In quali parti del corpo e dove si esegue una iniezione intramuscolare?
- Com’è fatta una siringa e come si usa correttamente?
- Differenza tra dolore acuto, cronico, persistente ed episodico con esempi
- Differenza tra dolore somatico e psicosomatico
- Dolore: come si misura? La scala visiva e numerica verbale
- Dolore: quando chiamare il medico e cosa riferirgli
- Dolore: esistono esami specifici per rilevarlo?
- Meglio Aspirina o Ibuprofene?
- Supposta rettale: vantaggi e svantaggi rispetto ad altre vie di somministrazione
- Cosa succede ad una supposta dopo averla inserita? Come funziona?
- Supposte di glicerina: come usarle in bambini, adulti, gravidanza
- Come mettere facilmente una supposta a neonati, bambini, adulti
- Si possono tagliare o spezzare le supposte rettali?
- Lingua bianca, impastata, spaccata: cause e quando è pericolosa
- A che serve la Tachipirina (paracetamolo)?
- Gravidanza e allattamento: posso assumere Tachipirina, Ibuprofene e Co-Efferalgan? Quante compresse?
- Differenze tra Efferalgan e Co-Efferalgan
- Cardioaspirin 100mg: effetti indesiderati, a cosa serve, dosaggi (foglio illustrativo)
- Che significa somministrazione di un farmaco PER OS o PO?
- Via di somministrazione orale, per os: vantaggi e svantaggi
- Tachipirina, paracetamolo, Efferalgan: posologia, controindicazioni ed effetti collaterali
- Voltaren Emulgel (diclofenac): come usarlo, gravidanza ed effetti collaterali
- Arvenum: terapia di emorroidi e fragilità capillari
- Enterogermina per gonfiore, diarrea e dolori addominali: foglietto illustrativo
- Moment (ibuprofene): posologia, effetti collaterali, gravidanza, prezzo
- Rinazina spray nasale in bambini e adulti: posologia e prezzo
- Glicerolo Carlo Erba soluzione rettale e supposte: posologia ed effetti collaterali
- Triatec (ramipril): posologia, effetti collaterali [FOGLIETTO ILLUSTRATIVO]
- Lasix (furosemide): posologia, effetti collaterali [FOGLIETTO ILLUSTRATIVO]
- Norvasc (amlodipina): posologia, effetti collaterali [FOGLIETTO ILLUSTRATIVO]
- Augmentin (amoxicillina): posologia, effetti collaterali [FOGLIETTO ILLUSTRATIVO]
- Omeprazen (omeprazolo): posologia, effetti collaterali [FOGLIETTO ILLUSTRATIVO]
- Dibase (vitamina D): posologia, effetti collaterali [FOGLIETTO ILLUSTRATIVO]
- Torvast (atorvastatina): posologia, effetti collaterali [FOGLIETTO ILLUSTRATIVO]
- Differenza tra Gabapentin e Pregabalin
- Farmaco Lyrica (Pregabalin): indicazioni ed effetti collaterali
- Lyrica (Pregabalin): è un farmaco che fa ingrassare?
Lo Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o unisciti al nostro gruppo Facebook o ancora seguici su Twitter, su Instagram o su Pinterest, grazie!
