
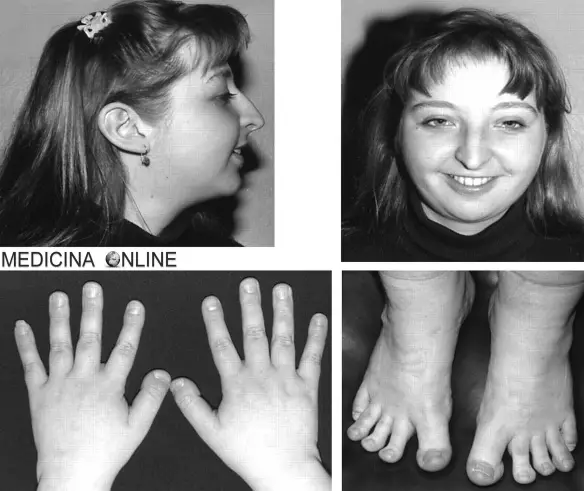
 La sindrome di Rubinstein-Taybi (in inglese “Rubinstein–Taybi syndrome“, da cui “RTS”) è una rara malattia genetica caratterizzata da bassa statura, difficoltà di apprendimento da moderata a grave (ritardo mentale), caratteristiche facciali distintive, pollici/alluci larghi e clinodattilia del 5° dito. Le persone con sindrome di Rubinstein-Taybi hanno un rischio maggiore di sviluppare tumori come leucemia e linfoma.
La sindrome di Rubinstein-Taybi (in inglese “Rubinstein–Taybi syndrome“, da cui “RTS”) è una rara malattia genetica caratterizzata da bassa statura, difficoltà di apprendimento da moderata a grave (ritardo mentale), caratteristiche facciali distintive, pollici/alluci larghi e clinodattilia del 5° dito. Le persone con sindrome di Rubinstein-Taybi hanno un rischio maggiore di sviluppare tumori come leucemia e linfoma.
Epidemiologia
La sindrome di Rubinstein-Taybi è molto rara: secondo alcune stime si verifica in circa 1 su 125.000-300.000 nascite. La sindrome di Rubinstein-Taybi non mostra distinzione tra sesso maschile e femminile, né per una specifica etnia.
Eponimo
La sindrome di Rubinstein-Taybi deve il suo nome al pediatra statunitense Jack Herbert Rubinstein ed al radiologo iraniano-statunitense Hooshang Taybi. che per primi fecero ricerche su di essa.
Cenni storici
La sindrome di Rubinstein-Taybi fu menzionata per la prima volta in modo non ufficiale in una rivista medica ortopedica francese nel 1957 dai medici greci Michail, Matsoukas e Theodorou. La rivista medica ha riportato un caso riguardante un bambino di sette anni con pollici radicalmente deviati/inarcati, naso lungo, ipotonia muscolare e sottosviluppo fisico e mentale. A questo punto il caso di studio menzionato dai medici greci era considerato un’anomalia per il fatto che non erano stati segnalati altri casi di bambini con queste specifiche caratteristiche fisiche e mentali. Questo è stato molto probabilmente il primo caso segnalato di RTS nella letteratura scientifica.
Il dottor Jack Herbert Rubinstein, un pediatra americano, ha riferito di aver avuto come paziente una bambina di tre anni con insoliti reperti facciali e digitali nel 1958. Allo stesso modo, quello stesso anno Rubinstein aveva valutato un altro bambino con caratteristiche simili, questa volta di sette anni. Avendo percepito una sorprendente somiglianza tra questi due casi non correlati, Rubinstein ha provato a distribuire foto e informazioni riguardanti questi due casi ad altre cliniche negli Stati Uniti dal 1959 al 1960. Rubinstein si è laureato alla Harvard Medical School e ha lavorato come direttore della Hamilton County Diagnostic Clinic per il Ritardato mentale. Ha lavorato in pediatria comportamentale e dello sviluppo per molti anni prima della scoperta di questa nuova sindrome.[citazione necessaria]
Nel 1961, il dottor Hooshang Taybi, un radiologo pediatrico iraniano-americano, riferì di aver valutato un bambino di tre anni che sembrava avere la stessa sindrome descritta da Rubinstein. Durante l’estate del 1963 il dottor Taybi riferì di aver valutato sette bambini con caratteristiche come pollici e alluci ampi, caratteristiche facciali “insolite” e disabilità intellettive – questi risultati apparvero nell’American Journal of Diseases of Children che documentava queste caratteristiche come una sindrome. Il Dr. Hooshang Taybi si è laureato alla Scuola di Medicina dell’Università di Teheran e ha lavorato per il Ministero della Salute. Più tardi nella sua carriera ha insegnato e praticato radiologia pediatrica in Oklahoma e Indiana.
Nel 1992 sono state identificate le prime anomalie genetiche che fungono da marker per la sindrome di Rubinstein-Taybi.
Cause
La sindrome di Rubinstein-Taybi è una malattia genetica causata da una mutazione o delezione nel gene CREBBP localizzato sul cromosoma 16 (mappato in p13.3) e/o del gene EP300 localizzato sul cromosoma 22. Il cromosoma specifico interessato da una mutazione determina il tipo di sindrome di Rubinstein-Taybi che può verificarsi. La mutazione del gene CREBP sul cromosoma 16 dà origine alla prima forma di RTS (la più comune). Mentre una mutazione del gene EP300 sul cromosoma 22 è caratteristica della seconda forma di RTS (la più rara). La sindrome di Rubinstein-Taybi è talvolta ereditata con trasmissione autosomico dominante (vedi il prossimo paragrafo), ma molte volte si verifica come un evento de novo (sporadico, quindi non ereditato).
Il gene CREBBP (cromosoma 16) produce una proteina che aiuta a controllare l’attività di molti altri geni. La proteina, chiamata proteina legante CREB, svolge un ruolo importante nella regolazione della crescita e della divisione cellulare ed è essenziale per il normale sviluppo fetale. Se una copia del gene CREBBP viene eliminata o mutata, le cellule producono solo la metà della normale quantità di proteina legante CREB. Una riduzione della quantità di questa proteina interrompe il normale sviluppo prima e dopo la nascita, portando ai segni e sintomi della più comune forma della sindrome di Rubinstein-Taybi.
Le mutazioni nel gene EP300 (cromosoma 22) sono responsabili di una piccola percentuale di casi di sindrome di Rubinstein-Taybi. Queste mutazioni provocano la perdita di una copia del gene in ciascuna cellula, che riduce della metà la quantità di proteina p300. Alcune mutazioni portano alla produzione di una versione molto breve e non funzionale della proteina p300, mentre altre impediscono a una copia del gene di produrre qualsiasi proteina. Sebbene i ricercatori non sappiano come una riduzione della quantità di proteina p300 porti alle caratteristiche specifiche della sindrome di Rubinstein-Taybi, è chiaro che la perdita di una copia del gene EP300 interrompe il normale sviluppo.
Sfortunatamente, in quasi il 40% dei casi, nessuno dei due geni, CREBBP o EP300, può essere implicato nella causa della malattia. In questi casi, non vi è alcuna mutazione sul cromosoma 16 né sul 22, lasciando molte altre domande ancora senza risposta. Tuttavia, una nuova ricerca mostra che circa il 55% dei casi coinvolge una mutazione nel gene CBP, suggerendo ulteriormente che questa malattia ha le sue origini nella mutazione dei meccanismi regolatori della trascrizione (Izumi, 2016).
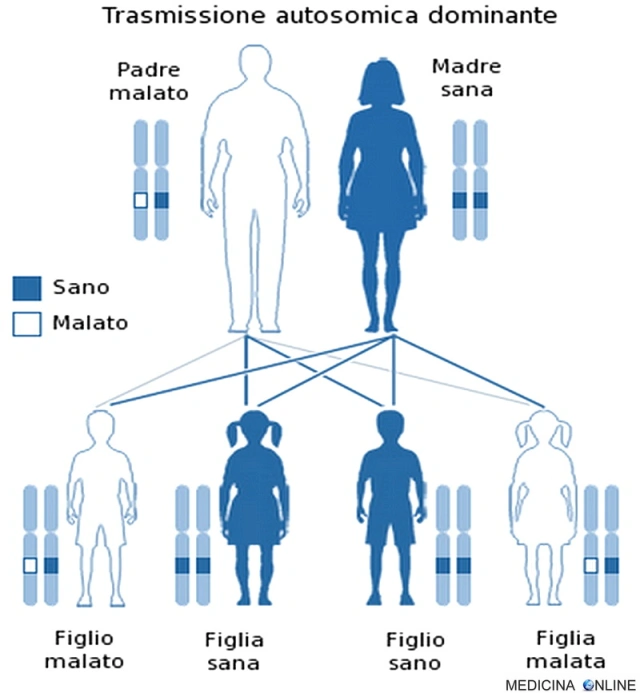
Trasmissione autosomica dominante
Una malattia è detta a trasmissione autosomica dominante quando basta una singola copia dell’allele difettoso per far sì che la malattia si esprima, a prescindere dal sesso (basta un solo genitore malato). Il figlio di un individuo affetto ha la probabilità del 50% di essere affetto, cioè 1 figlio su 2 è malato e può trasmettere a sua volta la malattia alla metà dei suoi figli. In questo caso non può esistere un “portatore sano” (cosa che invece si può verificare nella trasmissione autosomica recessiva): chi possiede l’allele alterato, ha la patologia, mentre chi non lo possiede è sano. Di conseguenza da due genitori sani nascono il 100% di figli sani, mentre se entrambi i genitori sono malati allora si avranno il 100% di figli malati.
Sintomi e segni
Le caratteristiche tipiche del disturbo includono:
- disabilità mentale generalmente moderata o grave,
- bassa statura,
- ritardo nella crescita,
- microcefalia (testa piccola),
- ostruzione dei dotti lacrimali,
- strabismo,
- blefaroptosi,
- stenosi polmonare,
- iperlassità articolare,
- pollici ed alluci larghi,
- clinodattilia (curvatura) del mignolo,
- ipotonia muscolare,
- reflusso gastroesofageo,
- coartazione aortica,
- formazione di cheloidi,
- criptorchidismo nei maschi,
- deficit di memoria e di attenzione,
- scarsa coordinazione motoria,
- difficoltà a deambulare,
- facies insolite che coinvolgono gli occhi, il naso e il palato
- anomalie cardiache,
- anomalie scheletriche,
- compromissione uditiva,
- scoliosi,
- anomalie del tratto urinario (inclusi i reni),
- problemi dentali
- problemi del linguaggio.
Non tutti i pazienti soffriranno di tutti o più sintomi e non tutti i pazienti sperimenteranno gli stessi sintomi con la medesima gravità.
Complicanze
L’anestesia può essere pericolosa in questi pazienti: in alcuni casi, gli individui con sindrome di Rubinstein-Taybi possono avere complicazioni (ad esempio distress respiratorio e/o aritmie cardiache) associate al miorilassante succinilcolina e altri determinati farmaci usati nelle anestesie. Qualsiasi situazione che richieda la somministrazione di anestesia o succinilcolina (ad esempio procedure chirurgiche) deve essere attentamente monitorata da professionisti qualificati.
I bambini possono avere un tasso più alto di anomalie cardiache, in particolare anomalie della conduzione elettrica dell’impulso cardiaco, che possono causare risultati imprevisti con i farmaci cardioattivi. Le persone con sindrome di Rubinstein-Taybi hanno anche un rischio maggiore di sviluppare tumori come leucemia e linfoma.
Uno studio del 2009 ha rilevato che i bambini con sindrome di Rubinstein-Taybi avevano maggiori probabilità di essere in sovrappeso e avere una breve capacità di attenzione, stereotipie motorie e scarsa coordinazione. Lo studio ha ipotizzato che il gene CREBBP identificato compromettesse l’apprendimento delle abilità motorie. Altre ricerche hanno mostrato un legame con il deficit di memoria a lungo termine.
Diagnosi
La sindrome di Rubinstein-Taybi viene sospettata in base all’anamnesi ed all’esame obiettivo e diagnosticata con test genetico che identificata una variante del gene CREBBP e/o del gene EP300. La sindrome esibisce un modello di ereditarietà autosomica dominante, ma alcuni casi documentati mostrano individui eterozigoti che esibiscono mosaicismo germinale. Spesso la malattia viene confusa con altre patologie che provocano ritardo mentale e della crescita. Le anomalie alle dita possono confondere il medico, che potrebbe pensare alla presenza di macrodistrofia lipomatosa.
Terapia
Non esiste un trattamento esistente che inverta o curi a monte la sindrome di Rubinstein-Taybi, esistono, tuttavia, modi per gestire e ridurre i sintomi per i pazienti. I pazienti con sindrome di Rubinstein-Taybi soffrono di una vasta gamma di sintomi, quindi vengono indirizzati a diversi specialisti che si concentrano ognuno su un sintomo specifico. Non esiste quindi un solo specialista che si occupa della sindrome. Ad esempio, i pazienti avranno bisogno di un chirurgo ortopedico e di un fisioterapista che si occuperanno di anomalie scheletriche, della crescita, della postura e del movimento, e avranno bisogno di un cardiologo se soffrono di anomalie cardiache o da un dentista se soffrono di anomalie dentali. In caso di obesità, un dietologo è raccomandato. Gli individui che soffrono di sviluppi cognitivi di solito hanno bisogno di programmi di educazione speciale e di logopedia. Sono necessari controlli e monitoraggio regolari relative alle anomalie cardiache, dentali, uditive e renali, soprattutto se sono gravi. La consulenza genetica è raccomandata anche per le persone colpite e le loro famiglie.
Leggi anche:
- Ritardo mentale (disabilità intellettiva) nei bambini lieve, moderato, grave: si guarisce?
- Sindrome dell’idiota sapiente: cause, caratteristiche e sintomi
- Sindrome del tramonto o del crepuscolo: cause, sintomi e cura
- Che cos’è l’intelligenza umana: definizione, significato e psicologia
- Quoziente d’intelligenza: valori, significato, test ed ereditarietà
- Problem solving: cos’è, caratteristiche, tecniche, fasi ed esempi
- Sindrome di Rett: cause, sintomi, tipi, diagnosi, stadi, cure, morte
- Deficit di attenzione: quando un bambino è iperattivo, che fare?
- Sindrome di Tourette: cause, sintomi, diagnosi e trattamento
- Sindrome di Tourette: si può guarire definitivamente? Come si guarisce?
- Bullismo e prepotenza a scuola: il disturbo della condotta
- Litigi e mancato rispetto delle regole: il disturbo oppositivo provocatorio
- Ansia e paura di andare a scuola: la fobia scolare, sintomi e cure
- L’alterazione del sistema dell’attaccamento: il disturbo dell’attaccamento
- Com’è fatto il cervello, a che serve e come funziona la memoria?
- Il disagio psicologico nel bambino con dislessia
- Epilessia: riconoscere in tempo l’arrivo di una crisi e come comportarsi
- Epilessia infantile: come comportarsi col proprio figlio?
- Si può morire di epilessia?
- Demenza senile: cause, sintomi, decorso e cure
- Terapia occupazionale: caratteristiche, attività, obiettivi, dispositivi
- Disturbi cognitivi congeniti e acquisiti: ritardo mentale e demenza
- Riabilitazione cognitiva: Token Economy e tecniche per incrementare comportamenti adeguati
- Riabilitazione cognitiva: tecniche per incrementare comportamenti adeguati non presenti
- Riabilitazione cognitiva: tecniche per incrementare attenzione, memoria, linguaggio
- Disturbi pervasivi dello sviluppo (disturbi dello spettro autistico)
- Autismo: definizione, cause, sintomi, diagnosi e cure
- Sindrome di Asperger in bambini ed adulti: primi sintomi, terapie
- Disturbo disintegrativo dell’infanzia (demenza infantile): cause, sintomi e terapie
- Disturbo pervasivo dello sviluppo non altrimenti specificato
- Sindrome di Down: rischio di avere un figlio affetto
- Quanti cromosomi ha chi è affetto da Sindrome di Down?
- Le malattie genetiche più diffuse al mondo
- Dislessia: cos’è, come riconoscerla, come affrontarla e superarla
- Persone famose con la Sindrome di Asperger
- Vostro figlio soffre di autismo? I primi segnali per capirlo e come comportarsi con lui
- Disturbi specifici di apprendimento (DSA): definizione, cause, sintomi, cure
- Disgrafia: esempi, come riconoscerla precocemente, test, rimedi
- Disortografia: cause, come riconoscerla, esempi, rimedi, si guarisce?
- Differenze tra disgrafia e disortografia
- Discalculia: significato, tipi, sintomi, diagnosi e terapia
- Disprassia a scuola: sintomi, esercizi, si guarisce?
- Disturbo specifico della compitazione: significato, sintomi, cure
- Disturbo specifico del linguaggio: sintomi, classificazione, si guarisce?
- Ritardo semplice di linguaggio: definizione e classificazione
- Balbuzie e disfluenze: significato, cause, sintomi, rimedi
- Oligofrenia e sindrome oligofrenica: cause, sintomi, diagnosi, cura
- Makaton per bambini con disturbo specifico del linguaggio
- Braille alfabeto e numeri in italiano: come impararlo
- Lingua dei segni italiana: cos’è, come impararla, alfabeto, esempi
- LAD (Language Acquisition Device) di Noam Chomsky
- Le Tavole di Sviluppo di Kuno Beller
- Schizofrenia: sintomi iniziali, violenza, test, cause e terapie
- Differenza tra morbo di Alzheimer, demenza senile, vascolare e reversibile
- Com’è fatto il cervello, a che serve e come funziona la memoria?
- Sindrome di Turner: cariotipo, cause, sintomi e segni caratteristici
- Sindrome di Klinefelter: cariotipo, cause, sintomi e cura
- Sindrome di Down: cause, sintomi in gravidanza e nei neonati
- Sindrome di Noonan: cause, sintomi nel neonato, aspettative di vita
- Sindrome di Bloom: cause, sintomi, diagnosi e terapia
- Differenza tra psicologo e psicoterapeuta
- Differente approccio di psicologo, psicoterapeuta e psichiatra
- Differenze tra le varie scuole di psicoterapia: quale la più efficace?
- Disturbo ossessivo-compulsivo: ripetere, ripetere e ripetere ancora all’infinito un gesto. Differenze col disturbo di personalità ossessivo-compulsivo
- Fibrosi cistica polmonare: cos’è, sintomi in neonati e bambini, cure
- Malattia di Huntington: cos’è, ereditarietà, come si trasmette, età di insorgenza
- Morbo di Parkinson: cause, sintomi, decorso, terapie
- Morbo di Alzheimer: cause, sintomi, decorso, terapie
- Tumore al cervello: operato mentre suona la chitarra e canta Yesterday
- Differenza tra morbo di Alzheimer e morbo di Parkinson: sintomi comuni e diversi
- Sclerosi laterale amiotrofica (SLA): cause, sintomi, diagnosi e prognosi
- Sclerosi multipla: cause, sintomi, diagnosi e prognosi
- Differenze tra sclerosi laterale amiotrofica e sclerosi multipla
- Atrofia muscolare progressiva: cause, sintomi, cura, aspettativa di vita
- Disturbi cognitivi congeniti e acquisiti: ritardo mentale e demenza
Dott. Emilio Alessio Loiacono
Medico Chirurgo
Direttore dello Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o unisciti al nostro gruppo Facebook o ancora seguici su Twitter, su Instagram, su YouTube, su LinkedIn, su Tumblr e su Pinterest, grazie
