
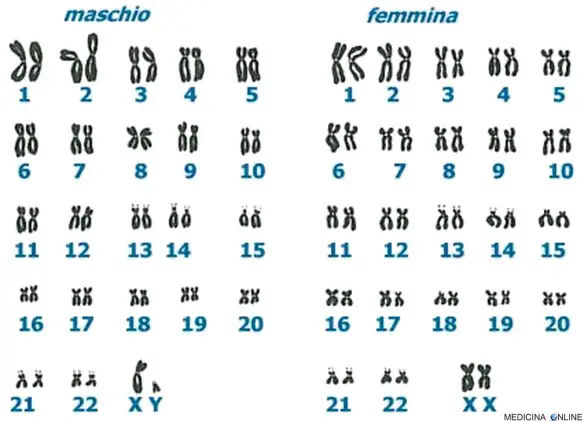
Nella sindrome di Edwards il cromosoma 18 è presente in tre copie e non in due
La sindrome di Edwards (anche chiamata “trisomia 18” da cui l’acronimo “T18”; in inglese “Edwards syndrome“) è una rara malattia genetica causata dalla presenza di un cromosoma 18 in più nel cariotipo: in pratica sono presenti tre cromosomi 18 invece che due, da cui il nome “trisomia 18”. Si tratta di una aneuploidia. La presenza di un cromosoma 18 in soprannumero è quasi sempre dovuta ad una non disgiunzione durante la meiosi. Le caratteristiche più comuni del bimbo affetto sono ritardo della crescita prenatale e postnatale, aspetto emaciato, facies caratteristica, anomalie degli arti, malformazioni viscerali, ipotrofia dei muscoli, microcefalia con cranio stretto, dolicocefalia, microretrognazia, ipertelorismo ed orecchie angolate a disegno semplice. La maggioranza dei casi si associa alla trisomia 18 libera. La trisomia 18 in mosaico è stata osservata in alcuni pazienti che presentano un quadro clinico che varia dalla forma classica di trisomia 18 a fenotipo normale, in rapporto al numero delle cellule trisomiche presenti nei tessuti. Il fenotipo della trisomia 18 è dovuto alla presenza di tre copie della regione 18q11-q12. Il rischio di ricorrenza di trisomia (13, 18 e 21) nelle famiglie con un caso indice è circa 1%. Tuttavia, nelle famiglie nelle quali la trisomia 18 è causata da una traslocazione, il rischio di ricorrenza è maggiore, se un genitore è portatore di una traslocazione bilanciata.
Cenni storici
La sindrome di Edwards deve il suo nome a John Hilton Edwards, che per primo la descrisse nel 1960.
Epidemiologia
La sindrome di Edwards si verifica in circa un caso su 6000-8000 nati vivi, tuttavia più feti sono colpiti dalla sindrome, poiché la maggior parte dei casi diagnosticati in fase prenatale (circa il 95%) morirà in utero prima della nascita per aborto spontaneo. Il tasso di sopravvivenza è più alto nelle femmine rispetto ai maschi: tra i feti affetti è quindi statisticamente più frequente che le femmine siano partorite. La sindrome di Edwards è la seconda più comune trisomia autosomica compatibile con la vita, dopo la sindrome di Down. Circa l’80% delle persone colpite è di sesso femminile. L’incidenza aumenta con l’aumentare dell’età della madre: l’età media materna per concepire un bambino con questo disturbo è di 32 anni e ½.
Cause e fattori di rischio
La sindrome di Edwards è causata da un’anomalia cromosomica caratterizzata dalla presenza di una copia extra del materiale genetico del cromosoma 18, sia totalmente (trisomia 18) o parzialmente (ad esempio a causa di una traslocazione). Il cromosoma supplementare solitamente compare prima del concepimento. Gli effetti della copia extra variano molto, a seconda del quantitativo di materiale genetico del cromosoma sovrannumerario. Un ovulo o uno spermatozoo sani contengono 23 cromosomi singoli che insieme formano una cellula normale del cariotipo umano con 46 cromosomi. Errori numerici possono insorgere in una delle due divisioni della meiosi e causare il fallimento della disgiunzione di una coppia nelle cellule figlie. Ciò si traduce in un cromosoma in più, rendendo il numero di totale di cromosomi a 24 invece di 23. La fecondazione di un uovo o l’inseminazione di sperma che contiene un extra cromosoma porta al crearsi di una situazione di trisomia, cioè la presenza di tre copie di un cromosoma invece che due. La trisomia 18 (47,XX,18) è causata da un evento di non disgiunzione meiotica. Con la non disgiunzione, un gamete (vale a dire, uno spermatozoo o una cellula uovo) nasce con una copia extra del cromosoma 18, il gamete ha quindi 24 cromosomi. Quando viene combinato con il gamete normale dall’altro genitore, l’embrione possiederà 47 cromosomi, con tre copie del cromosoma 18. In una piccola percentuale di casi, circa il 10-15%, solo alcune cellule dell’organismo hanno una copia extra del cromosoma 18, con il risultato che si ha una popolazione mista di cellule con un diverso numero di cromosomi. Tali casi sono a volte chiamati sindrome di Edwards con mosaico genetico. Molto raramente, un pezzo del cromosoma 18 si attacca a un altro cromosoma (traslocazione) prima o dopo il concepimento. Gli individui affetti hanno due copie del cromosoma 18, più materiale extra di un ulteriore cromosoma 18 attaccato ad un altro cromosoma. Con una traslocazione, una persona ha una trisomia parziale per il cromosoma 18 e le anomalie sono spesso meno gravi rispetto alla tipica sindrome di Edwards. La traslocazione si ha nel 5%-10% dei casi.
Sintomi e segni
La sindrome di Edwards si manifesta numerose caratteristiche. Quelle più comuni nel bimbo affetto sono:
- ritardo della crescita prenatale e postnatale;
- ritardo mentale;
- aspetto emaciato;
- facies caratteristica;
- anomalie degli arti;
- difficoltà di alimentazione (scarsa suzione);
- difficoltà di respirazione;
- artrogriposi (contratture articolari multiple);
- malformazioni viscerali congenite multiple;
- ipotrofia dei muscoli;
- sterno corto;
- mani serrate;
- cisti dei plessi corioidei;
- pugno chiuso con indice sovrapposto al medio (a uncino);
- piede equino;
- criptoorchidismo (ritenzione dei testicoli nei maschi).
Tra le malformazioni si osserva spesso:
- malformazioni del tratto genito-urinario (idronefrosi ed agenesia mono-bilaterale del rene);
- difetti cardiaci strutturali (oltre il 90% dei casi), come difetto del setto ventricolare, difetto interatriale e pervietà del dotto di Botallo;
- intestino sporgente al di fuori del corpo (onfalocele);
- malformazioni ano-rettali;
- atresia esofagea;
- aplasia del radio;
- spina bifida;
- anencefalia;
- oloprosencefalia.
La facies caratteristica include le seguenti condizioni:
- retrognazia;
- mandibola anormalmente piccola (micrognazia);
- palatoschisi;
- naso all’insù;
- occhi ampiamente distanziati (ipertelorismo oculare);
- microftalmia;
- coloboma;
- abbassamento delle palpebre superiori (ptosi);
- orecchie malformate (angolate a disegno semplice);
- microcefalia (test piccola) con cranio stretto;
- dolicocefalia.
Le anomalie dei piedi comprendono il piede equino-varo e/o a piccozza, con sovrapposizione delle dita (il V e il II dito ricoprono il IV e il III).
Diagnosi prenatale
La trisomia 18 può essere sospettata durante la gravidanza con l’ecografia e può essere confermata con l’analisi del cariotipo fetale (amniocentesi o villocentesi). Anche i marcatori sierici (utilizzati per la diagnosi di trisomia 21 o sindrome di Down) possono essere alterati. E’ possibile fare una stima del rischio che il feto sia affetto da trisomia 18 o da altre anomalie come la sindrome di Down, grazie alla valutazione dei fattori di rischio e ad una serie di esami assolutamente sicuri sia per la madre che per il feto tra cui duo test, tri test, quad test, translucenza nucale, test combinato, test integrato e test integrato sierico. All’ecografia, le caratteristiche più comuni sono le anomalie cardiache, seguite da quelle del sistema nervoso centrale. All’ecografia si nota ritardo di crescita, presenza di malformazioni e di cisti multiple dei plessi corioidei. L’anomalia intracranica più comune è la presenza di cisti del plesso coroide, sacche di fluido sul cervello. A volte si osserva eccesso di liquido amniotico. Tra gli esami più invasivi, ma anche più sensibili, ci sono invece amniocentesi e villocentesi (esame dei villi coriali), che sono eseguiti in genere tra la 9ª e la 18ª settimana di gestazione ed hanno un rischio abortivo rispettivamente dello 0.1-0.8% e dell’1.8%. In virtù dei rischi di aborto connessi ad amniocentesi e villocentesi, esse non vengono proposte di routine a tutte le donne incinte, bensì solo a quelle giudicate ad elevato rischio di anomalie cromosomiche a causa della presenza di numerosi fattori di rischio, tra cui:
- età della paziente superiore ai 35 anni;
- genitori portatori di alterazioni cromosomiche (traslocazioni, inversioni, aneuploidie) o gravi malattie genetiche (talassemie, fibrosi cistica etc.) già note o rilevate tramite test genetico pre-concepimento;
- feto considerato ad alto rischio di anomalie in base ai risultati di duo test, test combinato, tri test, quad test, translucenza nucale, test combinato, test integrato e/o test integrato sierico;
- aumentato spessore della translucenza nucale;
- presenza di difetti fetali individuati con l’ecografia;
- uno o più figli precedenti affetti da anomalia cromosomica;
- malattie infettive (ad esempio da citomegalovirus o da parvovirus B19);
- infiammazioni in utero (l’esistenza di un’infezione endoamniotica è anche causa di diverse patologie che possono impedire il corretto svolgimento della gravidanza.
Terapia
Non esiste una terapia che possa invertire l’esito della sindrome nel feto. Dopo la nascita, la presa in carico è fondamentalmente solo di supporto. In alcuni casi il trattamento chirurgico delle malformazioni riesce a migliorare la prognosi, che però è spesso infausta. Terapia fisica, occupazionale e del linguaggio aiuterà le persone con sindrome di Edwards a raggiungere il loro pieno potenziale di sviluppo, che però è purtroppo inferiore alla media.
Prognosi e mortalità
La sindrome ha un tasso molto basso di sopravvivenza, derivante da anomalie cardiache, malformazioni renali e altri problemi negli organi interni. La maggior parte dei feti con la sindrome muore prima della nascita. La metà dei bambini con questa condizione non sopravvive oltre la prima settimana di vita. La durata della vita media è di 5-15 giorni. Il 92% dei bambini muore nel primo anno di vita a causa delle complicazioni cardiache, renali o neurologiche o delle infezioni ricorrenti. Circa l’8% dei bambini sopravvive per più di 1 anno. L’1% dei bambini vive fino a 10 anni: ciò generalmente avviene nei casi meno gravi della sindrome di Edwards, ovvero quando vi è un mosaico genetico. Sono stati osservati rari pazienti sopravvissuti in alcuni casi fino ad età adulta, soprattutto in presenza di trisomia in mosaico o trisomia parziale (secondaria a traslocazione). La maggioranza dei pazienti non in mosaico sviluppa un’autonomia molto limitata (assenza del linguaggio e della deambulazione). Nei pazienti adulti, il ritardo mentale ed i deficit senso-motori costringono il soggetto a dover avere assistenza continua.
Leggi anche:
- Sindrome di Down: cause, sintomi in gravidanza e nei neonati
- Sindrome di Patau (trisomia 13): cause, sintomi, diagnosi, cure
- Malattia di Huntington: cos’è, ereditarietà, come si trasmette, età di insorgenza
- Anemia falciforme: cosa significa, cause, sintomi e cure
- Sindrome di Turner: cariotipo, cause, sintomi e segni caratteristici
- Sindrome di Klinefelter: cariotipo, cause, sintomi e cura
- Differenze tra la distrofia muscolare di Duchenne e di Becker
- Fibrosi cistica polmonare: cos’è, sintomi in neonati e bambini, cure
- Consulenza genetica, diagnosi prenatale, amniocentesi, esame dei villi coriali, tri test, screening GUIDA COMPLETA
- Test genetici diagnostici, screening neonatale esteso e test genetici prenatali
- Ecografie 3D e 4D: a cosa servono e quali sono le differenze con l’ecografia standard?
- Duo test in gravidanza: settimana, risultati, rischi, procedura, costo
- Tri test in gravidanza: settimana, risultati, rischi, procedura, costo
- Quad test in gravidanza: settimana, risultati, rischi, procedura, costo
- Translucenza nucale in gravidanza: a cosa serve, quando si fa, a chi è consigliata?
- Test combinato (duo test più translucenza nucale) in gravidanza
- Test integrato in gravidanza: a cosa serve, quando si fa, a chi è consigliato?
- Test integrato sierico: a cosa serve, quando si fa, a chi è consigliato?
- Differenze tra duo test, tri test, quad test, translucenza nucale, amniocentesi, villocentesi, test combinato e integrato
- Analisi del cariotipo, cariotipo normale e patologico, amniocentesi, villocentesi
- Amniocentesi precoce e tardiva: settimana, risultati, rischi, procedura, dolore, costo
- Analisi dei villi coriali (villocentesi): settimana, risultati, rischi, procedura, dolore, costo
- Differenze tra amniocentesi e villocentesi (prelievo dei villi coriali) vantaggi, svantaggi
- Cos’è un gene ed a che serve?
- Cos’è un cromosoma ed a che serve?
- Cosa sono gli alleli ed a che servono?
- Differenza tra genetica ed epigenetica
- Differenza tra genetica e genomica
- Differenza tra malattia genetica, ereditaria e congenita
- Differenza tra malattia autosomica dominante e recessiva con esempi
- Differenza tra dominanza semplice, incompleta e codominanza
- Differenza tra allele dominante e recessivo
- Differenza tra portatore sano e malato in genetica
- Cosa significa “portatore sano” in genetica e nelle infezioni?
- Differenza tra omozigote ed eterozigote
- Differenza tra genotipo e fenotipo
- Le 10 malattie genetiche più diffuse al mondo
- Ecografie 3D e 4D: a cosa servono e quali sono le differenze con l’ecografia standard?
- Per abortire serve il consenso dei genitori? Aborto per minorenni e interdette
- Aborto: entro quanto è legale?
- Aborto volontario: decide la madre, il padre o entrambi? E se la donna è minorenne?
- Dove abortire? Il medico è costretto a praticare l’aborto? Cos’è l’obiezione di coscienza?
- Raschiamento: è doloroso, quando avere rapporti, perdite e nuova gravidanza
- Aborto spontaneo: quali sono le cause ed i sintomi precoci?
- Aborto spontaneo: cos’è, sintomi iniziali e come riconoscerlo
- Differenza tra aborto spontaneo, completo, incompleto, ritenuto
- Differenza tra aborto spontaneo, parto prematuro e “nato morto”
- Differenza tra aborto interno e spontaneo
- Differenza tra aborto spontaneo, provocato, volontario e terapeutico
- Differenza tra aborto e interruzione di gravidanza
- Differenza tra aborto e morte intrauterina fetale
- Differenza tra aborto e gravidanza biochimica
- Differenza tra aborto e raschiamento
- Spina bifida e difetti di chiusura del tubo neurale nel feto: trasmissione, prevenzione, diagnosi e cura
- Meningocele: cause, sintomi, diagnosi, cura e prevenzione
- Mielomeningocele, spina bifida e schisi vertebrale: complicanze, cura, prevenzione
- Encefalocele: cause, sintomi, diagnosi, cura e prevenzione
- Prendere il sole in gravidanza fa male al bambino?
- Estate e gravidanza: quale cibi devi preferire se sei in dolce attesa
- Quali creme solari sono più indicate in gravidanza?
- Gravidanza in estate: come ci si difende dal caldo e dall’afa?
- E’ possibile farsi il bagno al mare o in piscina in gravidanza?
- Cloasma gravidico: cos’è e come si previene in estate?
- Troppo sole in gravidanza “surriscalda” il liquido amniotico?
- Gravidanza: come indossare la cintura di sicurezza in auto?
- Gravidanza: è possibile viaggiare e prendere l’aereo o fa male al bimbo?
- Gravidanza e bambino podalico: manipolazione ed esercizi
- Parto cesareo: dopo quanto si possono avere rapporti sessuali?
- Dopo il parto: come recuperare l’intimità di coppia?
- Perché riprendere l’attività sessuale dopo il parto è così difficile?
- Parto naturale: dopo quanto si possono avere rapporti sessuali?
- Come fare gli esercizi di Kegel: la ginnastica pelvica per migliorare il piacere sessuale femminile e aiutare il parto
- A che serve la vitamina B12? L’importanza in gravidanza e allattamento
- Diabete gestazionale: cos’è e quali sono i rischi per il feto e la madre
- Ragazza è sterile per un tumore: sua madre partorisce un figlio per lei
- Depressione post parto: come riconoscere i primi sintomi e superarla
- Aumentare il ferro in modo naturale, specie in gravidanza
- Perdite bianche, gialle marroni in gravidanza: quando preoccuparsi e cosa fare?
- Acido folico (vitamina B9): a cosa serve, in quali alimenti trovarlo e perché è importante prima e durante la gravidanza
- Pubalgia in gravidanza: cause e rimedi del dolore all’osso pubico
- Pubalgia acuta e cronica: sintomi, esercizi e rimedi
- Differenza tra pube ed osso iliaco: anatomia e funzioni
- Differenza tra pube e inguine
- Masturbarsi in gravidanza fa male al bambino?
- Fare sesso in gravidanza fa male bambino?
- Rapporti sessuali in gravidanza: la guida trimestre dopo trimestre
- Gravidanza: cosa percepisce il bambino durante un rapporto sessuale?
- Gravidanza: è vero che il desiderio sessuale aumenta?
- Quali sono le posizioni sessuali consigliate in gravidanza?
- Differenza dei capezzoli e del seno in gravidanza
- Differenza dolore al seno da gravidanza e da ciclo mestruale
- Prevenire le ragadi al capezzolo: cosa fare prima del parto, cosa fare quando si sta allattando
- Le cose che non devi MAI dire ad una donna incinta, soprattutto se sei una donna
Dott. Emilio Alessio Loiacono
Medico Chirurgo
Direttore dello Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o unisciti al nostro gruppo Facebook o ancora seguici su Twitter, su Instagram o su Pinterest, grazie!
