
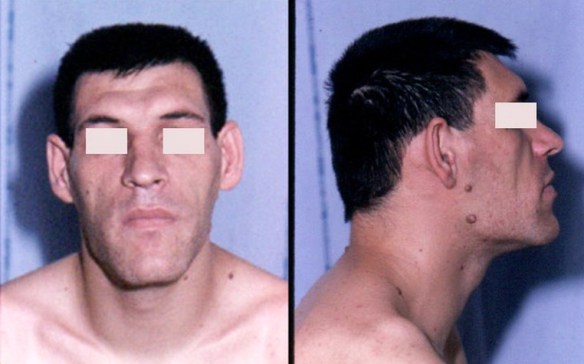
 L’acromegalia è una malattia cronica debilitante, caratterizzata dall’ingrossamento di piedi, mani, lingua e ossa del volto e dall’organomegalia, cioè dall’aumento di volume di organi interni (cuore, fegato, tiroide, intestino, rene). L’acromegalia ha un’incidenza annua di 3-4 casi per milione di abitanti, ma la prevalenza riportata di 40 casi per milione è in realtà sottostimata per la tardività della diagnosi e il misconoscimento sul territorio.
L’acromegalia è una malattia cronica debilitante, caratterizzata dall’ingrossamento di piedi, mani, lingua e ossa del volto e dall’organomegalia, cioè dall’aumento di volume di organi interni (cuore, fegato, tiroide, intestino, rene). L’acromegalia ha un’incidenza annua di 3-4 casi per milione di abitanti, ma la prevalenza riportata di 40 casi per milione è in realtà sottostimata per la tardività della diagnosi e il misconoscimento sul territorio.
Cause
L’acromegalia è determinata da un eccesso dell’ormone della crescita (GH) nell’età postpuberale e dal conseguente aumento del fattore di crescita insulinosimile 1 (IGF-1) secreto principalmente dal fegato in risposta al GH. Per approfondire: Ormone della crescita (GH) a che serve e da cosa è prodotto?
IMPORTANTE: se l’ipersecrezione di GH inizia prima della saldatura delle epifisi (età prepuberale), sono si verifica acromegalia, bensì gigantismo ipofisario. Per approfondire, leggi:
L’ipersecrezione ormonale è quasi sempre causata da un adenoma ipofisario GH secernente, mentre nei restanti rarissimi casi è dovuta ad un’ipersecrezione di GHRH di origine intracranica (adenomi ipotalamici a crescita lenta, choristomi, ganglioneuromi) o extracranica, più frequentemente carcinoidi bronchiali, raramente tumori neuroendocrinidel tratto gastrointestinale e carcinoidi timici. L’eccesso di GH può verificarsi anche in caso di uso di farmaci dopanti ed è tipico nel bodybuilding.
Per approfondire, leggi anche:
- Steroidi anabolizzanti: effetti collaterali fisici e psicologici in uomo e donna
- Mandibola GH, ormone della crescita e doping nello sport
- Ormone della crescita (GH): body building e doping in palestra
- Ormone della crescita (GH): effetti avversi nel body building e nello sport
- Io dico NO al doping in palestra, sempre e comunque
- Come riconoscere un atleta “natural” da un dopato in palestra
Sintomi
L’acromegalia è nota essere caratterizzata da una progressiva deformazione delle connotazioni somatiche e da un ampio range di complicanze sistemiche. I pazienti si accorgono che i lineamenti del volto diventano grossolani. Il processo inizia generalmente prima dei 40 anni, ma il riconoscimento della malattia può essere molto tardivo. Si manifesta ingrandimento delle estremità con aumento della misura delle scarpe, dei guanti, necessità di allargare gli anelli o il bracciale dell’orologio, aumento della misura del cappello. Le manifestazioni cliniche possono essere legate all’estensione locale della massa ipofisaria causando per lo più cefalea, difetti di campo visivo, paralisi dei nervi cranici e segni di ipopituitarismo. Molto raramente si assiste a rinoliquorrea e ostruzione nasale quando la crescita dell’adenoma è per lo più verso il basso. Ma la maggior parte delle complicanze sono legate all’ipersecrezione di GH causando organomegalia, alterazioni cardiovascolari, metaboliche, respiratorie, ossee.
La cefalea a localizzazione aspecifica (retroorbitaria, al vertice, occipitale), più accentuata al risveglio, risponde ai comuni antidolorifici. Dipende dallo stiramento della dura madre del diaframma della sella.
In caso di tumori estesi a livello sovraipofisario con compressione del forame di Monro o dell’acquedotto di Silvio si può arrivare ad avere ipertensione endocranica con i segni associati di vomito ed edema della papilla.
I difetti del campo visivo dipendono dalla localizzazione anatomica del chiasma, dal decorso dei nervi ottici e dal grado di compressione o stiramento degli stessi.
I difetti campimetrici, di solito, insorgono gradualmente con conseguenti meccanismi di adattamento, tipo modificazioni dei movimenti del capo per mantenere la visione. Questo ritarda la diagnosi. Nelle lesioni che coinvolgono il chiasma si manifesta emianopsia bitemporale che progredisce in senso orario a destra e in senso antiorario a sinistra. Generalmente l’acuità visiva è normale. La presenza di emianopsia può determinare difficoltà nella guida (nella visione laterale) e nel parcheggiare la macchina.
I pazienti lamentano inoltre difficoltà nell’esecuzione di compiti fini come infilare l’ago o tagliare le unghie. Si può manifestare inizialmente con una ridotta percezione dei colori.
L’atrofia della papilla è un segno tardivo. Si associa con importante perdita di acuità visiva.
La paralisi dei nervi cranici si manifesta quando il tumore presenta crescita laterale verso il seno cavernoso ove passano il III, IV, VI nervo cranico e la branca oftalmica e mascellare del V. I sintomi sono costituiti da ptosi palpebrale, diplopia o parestesie facciali.
L’ipopituitarismo è per lo più legato ad un danno da schiacciamento delle linee cellulari contigue all’adenoma. La linea cellulare più facilmente danneggiata è quella gonadotropino-secernente, seguita dalla GH secernente. Le più resistenti sono le linee tireotropino e corticotropino-secernenti. Non sempre tale ordine di perdita funzionale è rispettato.
Il deficit di gonadotropine si associa in epoca pre-puberale a mancato sviluppo o a blocco dello sviluppo puberale. In età adulta si manifesta con perdita di libido in ambedue i sessi ed irregolarità mestruali fino all’amenorrea nella femmina e disfunzione erettile nel maschio.
A questi si associano atrofia mammaria ed osteoporosi nella donna e riduzione di caratteri sessuali secondari e della massa muscolare ed osteoporosi nel maschio.
Il deficit di tireotropina si manifesta con sintomi meno marcati che nell’ipotiroidismoprimitivo: astenia, intolleranza al freddo e stitichezza.
Il deficit corticotropinico si manifesta anch’esso con astenia, anoressia, ipotensione ortostatica, perdita di peso, perdita della peluria nelle aree androgeno-dipendenti. Nell’ipocorticismo di origine centrale di solito la secrezione di aldosterone viene risparmiata. Di pari non si manifesta l’iperpigmentazione cutanea tipica del morbo di Addison.
Le altre manifestazioni cliniche dell’acromegalia dovute ad una ipersecrezione di GH comprendono:
- cardiomiopatia acromegalica;
- ipertensione;
- complicanze respiratorie;
- sindrome delle apnee del sonno;
- complicanze metaboliche;
- patologia tiroidea (gozzo multinodulare);
- manifestazioni osteoarticolari;
- tunnel carpale.
Diagnosi
La valutazione del paziente acromegalico prevede la valutazione basale e dopo test di stimolo dell’asse GH-IGF-1. Le valutazioni basali comprendono il dosaggio di GH e IGF-1. Un dosaggio random di GH inferiore a 0.4 mcg/l associato a livelli di IGF-1 nel range di normalità per sesso ed età e in assenza di altra patologia permette di escludere la diagnosi di acromegalia. Se uno o entrambi i parametri sono alterati, o vi è un forte sospetto di alterazione, è opportuno eseguire un test di soppressione dopo carico orale di glucosio (OGTT: oral glucose tolerance test) con 75 g di glucosio. Il test va effettuato al mattino a digiuno: dopo aver somministrato 75 g di glucosio si debbono eseguire prelievi per GH e glicemia ogni 30’ per almeno 2 ore. La mancata soppressione del GH sotto ad 1 mcg/l assieme a elevati livelli di IGF-1 per sesso ed età, indica presenza di patologia. Un aumento paradosso dei valori di GH durante il test non è indicativo di una maggior aggressività di malattia. Delle risposte falsamente positive si possono ritrovare nei soggetti adolescenti, diabetici, nell’anoressia nervosa, nell’insufficienza renale ed epatica, mentre falsi negativi (normale soppressione del GH) sono riscontrabili in quei pazienti acromegalici con bassi livelli circolanti di GH. Risulta abbastanza semplice l’interpretazione di questi dati se valutati anche i valori di IGF-1 e la clinica del paziente. Con l’introduzione di metodiche molto più sensibili per la misurazione del GH, è stato proposta una diminuzione dell’inibizione del cut-off del GH dopo OGTT. La soppressione del GH dopo OGTT assieme al dosaggio di IGF-1 sono ormai considerati i più importanti parametri per la diagnosi e il follow-up della patologia, perché si sono dimostrati essere molto sensibili e specifici e ben riproducibili.
In passato altri test venivano utilizzati per la diagnosi come il TRH, il GHRH, il GnRH, o la valutazione della secrezione spontanea giornaliera di GH, ma, in realtà, non aggiungono alcun tipo di informazione ai semplici test sopra proposti.
Poiché dal 50 al 70% dei pazienti acromegalici presenta alla diagnosi una risposta paradossa al test con TRH, questo parametro, potrebbe avere un senso diagnostico se utilizzato come parametro di confronto per eventuale persistenza di patologia nei pazienti sottoposti ad adenectomia ipofisaria.
Complemento diagnostico mediante radiologia
L’esame di prima scelta risulta essere ormai la RMN della regione ipotalamo ipofisariaeseguita in modo dinamico e con mezzo di contrasto. La metodica dinamica prevede l’esecuzione di sezioni in basale e subito dopo l’iniezione del mdc. Questa sfrutta il fatto che il circolo arterioso della parte sana dell’ipofisi è più efficiente e il tessuto sano si impregna più rapidamente rispetto a quello tumorale che appare così come un’area ipodensa/ipointensa. Parimenti il tessuto tumorale cede il contrasto più lentamente rispetto al tessuto sano e pertanto la lesione tumorale appare iperdensa/iperintensa nelle immagini tardive (ottenute dopo circa 20 min dall’iniezione del mdc).
La valutazione mediante RMN è importante anche per quanto concerne la valutazione della compressione del chiasma ottico e l’invasione del seno cavernoso. L’utilizzo della TAC è invece un completamento pre operatorio per una miglior valutazione del tessuto osseo.
Terapia
La terapia mira ad ottenere tre risultati principali:
- invertire la crescita del tumore
- diminuire la gravità dei sintomi
- diminuire l’alta mortalità
Come trattamento è previsto l’intervento chirurgico, e fra le varie possibilità l’operazione di scelta rimane la resezione transsfenoidale, il cui successo è molto alto, 80 ma anche 90% dei casi, mentre in attesa dell’operazione vengono somministrati analoghi della somatostatina, che fornisce molteplici effetti: da un lato riduce i sintomi offrendo sollievo ai pazienti e dall’altra diminuisce le masse tumorali interessate. I farmaci utilizzati sono:
- Octreotide acetato (analogo della somatostatina, scoperto nel 1970), molto più potente della somatostatina; la dose da somministrare è di 50 ug (3 volte al giorno) e, per allungarne la durata, viene modificato con il mannitolo in una formulazione nota come Octreotide LAR.
- Bromocriptina mesilato (dopamino-agonista), a dosi elevate per dare maggiori effetti, da 1,25 mg fino a 5 mg.
- Pegvisomant (antagonista dell’ormone della crescita),[17] che è stato testato anche su donne incinte ed è stato dimostrato trasmettere al nascituro il farmaco in parti talmente piccole da non risultare nocivo.[18]
Radioterapia
Si utilizza come terapia con un dosaggio molto elevato, normalmente 5000 cGy mentre la dose aumenta fino a raddoppiare se si utilizza il trattamento con protoni accelerati, ad una così elevata esposizione risultano diversi effetti indesiderati. Inoltre se il normale trattamento chirurgico non dovesse essere sufficiente si utilizzano delle nuove tecniche, introdotte recentemente, come il GKR (una terapia di radiochirurgia, dove si utilizza un coltello particolare a raggi gamma, per la persona comporta soltanto pochi effetti indesiderati.
Leggi anche:
- Asse ipotalamo-ipofisario: fisiologia e ormoni rilasciati
- Asse ipotalamo-ipofisi-gonade: funzionamento ed ormoni rilasciati
- Asse ipotalamo-ipofisi-testicolo: funzionamento ed ormoni rilasciati
- Asse ipotalamo-ipofisi-surrene: funzionamento ed ormoni rilasciati
- Asse ipotalamo-ipofisi-tiroide: funzionamento ed ormoni rilasciati
- Ipotalamo: anatomia, struttura e funzioni
- Differenze tra ipotalamo, ipofisi, neuroipofisi e adenoipofisi
- Patologie di ipotalamo e ipofisi
- Sindrome angio-osteoipertrofica (sindrome di Klippel-Trénaunay): cause, sintomi, diagnosi, cure
- Sindrome di Parkes Weber: cause, sintomi, diagnosi, terapie, prognosi
- Macrodistrofia lipomatosa: cause, sintomi, diagnosi, terapie
- Sindrome di Proteo: storia, cause, sintomi, diagnosi, terapie, complicanze
- Sindrome CLOVES: cause, sintomi, complicanze, diagnosi e terapia
- Sindrome di Sotos: cause, sintomi, diagnosi, terapia e prognosi
- Sindrome di Weaver-Smith: cause, sintomi, diagnosi, terapia e prognosi
- Gigantismo: cause, sintomi, diagnosi, terapia e prognosi
- Emimegaloencefalia: cause, sintomi, diagnosi, terapia e prognosi
- Diabete insipido: cause, diagnosi e trattamento
- Nanismo: sintomi, cura, cause, terapia, diagnosi e prevenzione
- Ipofisi (ghiandola pituitaria): anatomia, funzioni e ormoni secreti
- Sistema nervoso: com’è fatto, a che serve e come funziona
- Sistema nervoso simpatico: funzioni
- Sistema nervoso parasimpatico: funzioni
- Cos’è una ghiandola endocrina? A che servono gli ormoni ed il sistema endocrino?
- Feedback negativo ed omeostasi: spiegazione ed esempi
- Tiroide: dove si trova, com’è fatta e quali funzioni svolge?
- Ipotiroidismo: sintomi, diagnosi, cura farmacologica e consigli dietetici
- Tiroidite di Hashimoto e gravidanza
- Ormoni tiroidei: differenza T3 e T4, valori normali e patologici
- TSH alto, basso e valori normali: qual è il significato clinico?
- Ipertiroidismo: cause, cura, valori diagnosi, sintomi iniziali, conseguenze
- Ipertiroidismo nell’uomo: sintomi, conseguenze sulla libido, cure
- Morbo di Basedow: alimentazione, cura, occhi, si guarisce, rimedi
- Gozzo tiroideo: semplice, tossico, endemico, rimedi, intervento, immagini
- Gozzo tossico nodulare e multinodulare: sintomi, diagnosi e cura
- Alimenti gozzigeni (antitiroidei) vietati nelle alterazioni tiroidee
- Morbo di Plummer (adenoma tossico): sintomi, diagnosi e terapia
- Adenoma tiroideo: sintomi, diagnosi e trattamento
- Eutirox: quando si usa, dosaggio ed effetti collaterali (foglio illustrativo)
- Ipoparatiroidismo e ipocalcemia post chirurgici ed autoimmuni: sintomi e cure
- Tireotossicosi autoimmune, iatrogena, factitia, valori, cura
- Tiroidite acuta, cronica, autoimmune: sintomi e conseguenze
- Tiroidite di Hashimoto: esami, cura, conseguenze, dieta, guarire
- Tiroidite di De Quervain (subacuta): sintomi, dieta, si guarisce, è contagiosa?
- Tiroidite post partum: sintomi, fasi, diagnosi, cura e rischi
- Com’è fatto il cervello, a che serve e come funziona la memoria?
- Cervello maschile e femminile: quali sono le differenze?
- Sistema nervoso autonomo simpatico e parasimpatico: anatomia e funzioni
- Cos’è l’adrenalina ed a cosa serve?
- Adrenalina e “combatti o fuggi”: ecco cosa accade nel nostro corpo quando siamo terrorizzati
- L’altezza media italiana nel 2017 di maschi e femmine
- L’altezza media italiana 2017 da 1 a 19 anni di maschi e femmine
- L’altezza media mondiale nel 2017 di maschi e femmine [TABELLA]
- Altezza: quando si può parlare di nanismo o gigantismo
- Differenza tra midollo osseo e spinale
- Differenza tra sistema nervoso centrale e periferico: anatomia e funzioni in sintesi
- Quanto è alto l’uomo più alto del mondo?
- Quanto è alto l’uomo più basso del mondo?
- Charlotte, la bambina più piccola del mondo
- Tumore benigno e maligno del surrene: sintomi, diagnosi, cura
- Surrene: anatomia, funzioni e patologie in sintesi
- Asse ipotalamo-ipofisi-surrene: funzionamento ed ormoni rilasciati
- Ormone adrenocorticotropo o corticotropina (ACTH): cos’è e quali sono le sue funzioni
- Ormone di rilascio della corticotropina (CRH): produzione e funzioni
- Testosterone basso, alto, valori normali ed interpretazione
- Ormoni estrogeni: cosa sono e quali funzioni svolgono?
- Progesterone: cos’è, a cosa serve, valori e quali funzioni ha in gravidanza?
- Quando la donna ha troppi peli dove non dovrebbero essere: irsutismo, cause, trattamenti e differenze con ipertricosi
- Morbo di Addison: sintomi, immagini, terapia, mortalità, aspettative di vita
- Sindrome di Cushing: immagini, sintomi, diagnosi, cura, guarigione
- Feocromocitoma e surrenectomia: sintomi e conseguenze
- Surrenectomia: tecniche e conseguenze dell’asportazione del surrene
- Iperplasia surrenale congenita: tipi, sintomi, diagnosi, cura
- Iperplasia surrenale acquisita: cause, sintomi, terapia
- Iperandrogenismo femminile: significato, cause, sintomi e terapie
- Iperandrogenismo nell’uomo: significato, cause, sintomi e terapie
- Deficienza di ACTH e cortisolo (ipocortisolismo secondario)
- Deficienza di CRH e cortisolo (ipocortisolismo terziario)
- Malattia di Cushing: cause, sintomi, diagnosi, terapia
- Sindrome di Achard-Thiers
- Sindrome di Waterhouse-Friderichsen: cause e sintomi
Dott. Emilio Alessio Loiacono
Medico Chirurgo
Direttore dello Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o unisciti al nostro gruppo Facebook o ancora seguici su Twitter, su Instagram, su YouTube, su LinkedIn, su Tumblr e su Pinterest, grazie!
