
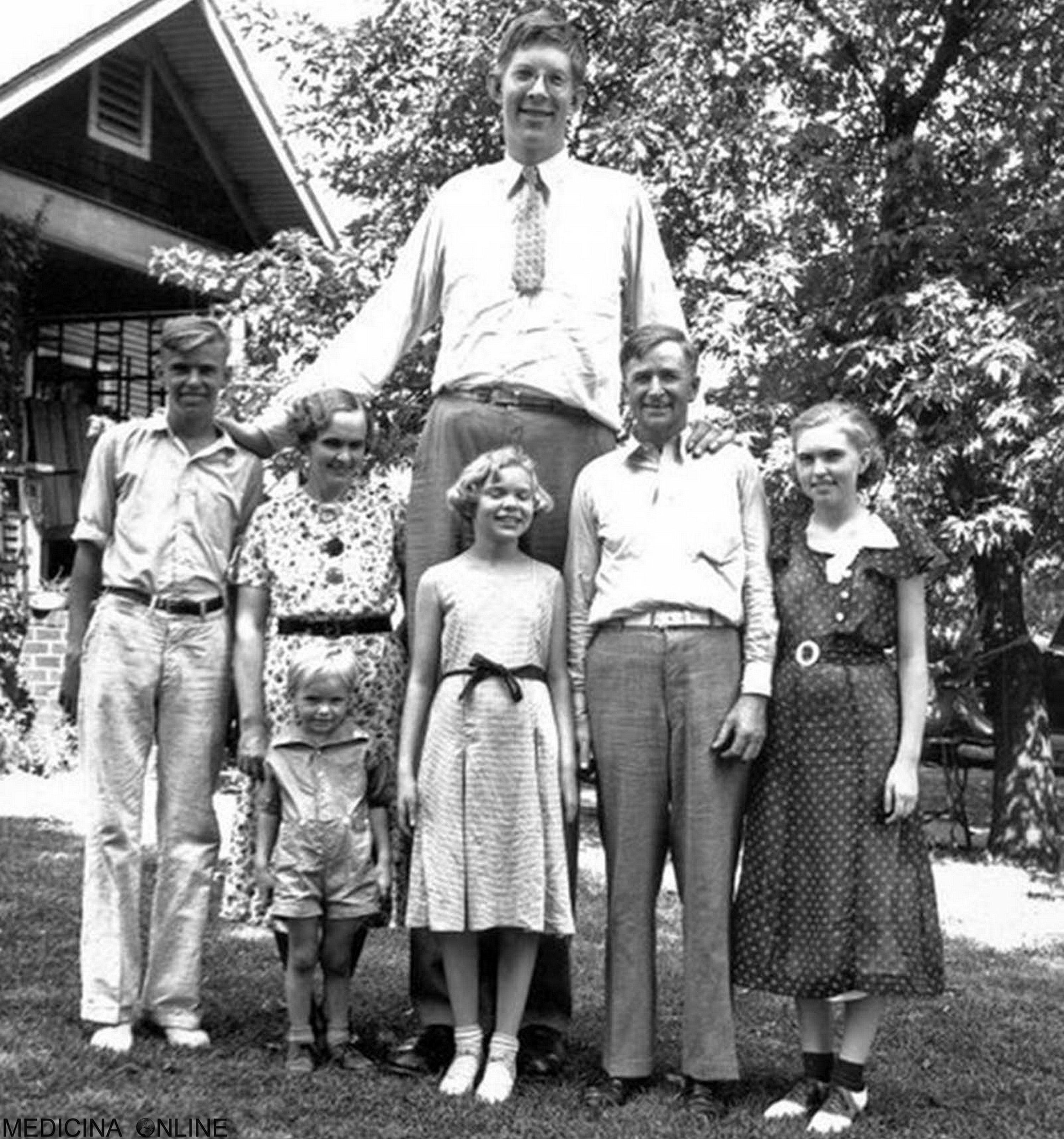 Il gigantismo (anche chiamato “macrosomia”, in inglese “gigantism”) è una patologia ormonale caratterizzata da un eccessivo accrescimento somatico delle strutture anatomiche del corpo. Il gigantismo non deve essere confuso con l’acromegalia, la forma adulta del disturbo, caratterizzata da un ingrossamento somatico specifico delle estremità e del viso.
Il gigantismo (anche chiamato “macrosomia”, in inglese “gigantism”) è una patologia ormonale caratterizzata da un eccessivo accrescimento somatico delle strutture anatomiche del corpo. Il gigantismo non deve essere confuso con l’acromegalia, la forma adulta del disturbo, caratterizzata da un ingrossamento somatico specifico delle estremità e del viso.
Etimologia
Il termine gigantismo deriva dal greco γίγας (gígas) che significa “gigante”.
Cause
Il gigantismo è causato da un’eccessiva esposizione dell’organismo all’ormone somatotropo, meglio noto come ormone della crescita o somatotropina (GH, da “growth hormone”), che si verifica durante l’età pre-puberale (prima della fusione della cartilagine delle ossa). Se l’organismo è esposto ad eccessivo ormone GH dopo la pubertà, si verifica invece acromegalia. L’eccesso di ormone della crescita che determina il gigantismo è praticamente quasi sempre causato da escrescenze ipofisarie (adenoma ipofisario, che determina gigantismo ipofisario), ma potrebbe essere anche causato dall’uso, prima della pubertà, di farmaci che aumentano il GH (ad esempio in caso di doping): in questo caso si parla di gigantismo iatrogeno.
Leggi anche:
- Ormone della crescita (GH) a che serve e da cosa è prodotto?
- Asse ipotalamo-ipofisario: fisiologia e ormoni rilasciati
Sintomi e segni
A causa delle quantità eccessive di ormone della crescita, i bambini raggiungono altezze ben al di sopra dei limiti normali. Un malato di gigantismo nell’età adulta può raggiungere anche altezze comprese tra i 240–274 cm, mantenendo però corrette le proporzioni corporee, a differenza dell’acromegalia, in cui l’ipersecrezione dell’ormone della crescita inizia dopo l’età puberale, e le proporzioni non sono più rispettate. La crescita staturale di un individuo è infatti permessa dalla presenza di piccole regioni, dette placche epifisarie, interposte tra l’epifisi e la diafisi delle ossa lunghe. Questi nuclei di accrescimento osseo producono cellule in grado di dividersi continuamente e sintetizzare nuovo tessuto cartilagineo, che viene poi ossificato a poco a poco. La saldatura delle placche epifisarie, che normalmente avviene intorno al 18º-20º anno di età, sancisce il definitivo arresto della crescita in altezza. L’età specifica di insorgenza del gigantismo varia a seconda dei pazienti e del sesso, ma l’età comune in cui iniziano a comparire i sintomi di una crescita eccessiva è stata trovata intorno ai 13 anni. Altre complicazioni di salute, come l’ipertensione, possono verificarsi in pazienti pediatrici con iper-secrezione dell’ormone della crescita. Caratteristiche più simili a quelle osservate nell’acromegalia possono verificarsi in pazienti che hanno visto un rapido aumento del GH in prossimità dell’adolescenza, poiché si stanno avvicinando alla fusione della cartilagine di accrescimento.
Diagnosi
La diagnosi avviene tramite esame obiettivo, esami di laboratorio, esami genetici ed esami di diagnostica per immagini (radiografie e TC). La valutazione dell’ipersecrezione dell’ormone della crescita non può essere esclusa con un singolo livello di GH normale dovuto alla variazione diurna. Tuttavia, un campione di sangue casuale che mostra GH marcatamente elevato è adeguato per la diagnosi di ipersecrezione di GH. Inoltre, un livello di GH normale elevato che non riesce a sopprimere con la somministrazione di glucosio è sufficiente anche per una diagnosi di ipersecrezione di GH. Il fattore di crescita insulino-simile-1 (IGF-1) è un eccellente test per la valutazione dell’ipersecrezione di GH. Non subisce variazioni diurne e sarà quindi costantemente elevato nell’ipersecrezione di GH e quindi nei pazienti con gigantismo. Un singolo valore normale di IGF-1 escluderà in modo affidabile l’ipersecrezione di GH.
Diagnosi differenziale
La diagnosi differenziale del gigantismo si pone nei confronti di varie patologie, tra cui:
- sindrome di Sotos;
- sindrome di Proteo;
- sindrome di Weaver-Smith;
- macrodistrofia lipomatosa;
- amartoma fibrolipomatoso;
- sindrome di Klippel-Trénaunay (sindrome angio-osteoipertrofica);
- neurofibromatosi di tipo 1;
- sindrome di Parkes Weber;
- sindrome di Rubinstein-Taybi;
- sindrome CLOVES;
- acromegalia.
Dal momento che il gigantismo è dovuto al cronico aumento della quota circolante di ormoni sessuali, in particolare di estrogeni, non sorprende che alcune forme di gigantismo siano tipiche della pubertà ritardata; si parla, in questi casi, di “gigantismo eunucoide”, caratterizzato da un aumento sproporzionato in lunghezza degli arti inferiori rispetto a quelli superiori. La pubertà precoce, al contrario, si accompagna ad un rapido accrescimento pre-termine, che si arresta tuttavia in maniera altrettanto rapida portando a deficit staturali in età adulta. Disturbi della pubertà sono abbastanza comuni anche nel gigantismo ipofisario. Un’altra forma, detta “gigantismo cerebrale” o “sindrome di Sotos”, in cui i livelli di GH circolanti sono normali; i bambini che ne sono affetti manifestano un eccessivo accrescimento corporeo durante i primi due o tre anni di vita, con notevole aumento della circonferenza cranica e sviluppo psicomotorio ritardato. Eccessi staturali si hanno anche nella sindrome di Marfan, una malattia genetica che colpisce il tessuto connettivo, ed in quella di Klinefelter, in cui l’alta statura è associata ad ipogonadismo, ginecomastia e scarso sviluppo pilifero.
Terapie
Non esiste una terapia che cura del tutto la sindrome: il trattamento limita i sintomi ed i livelli delle molecole che determinano iper-accrescimento. La terapia necessita un approccio multidisciplinare che coinvolge vari specialisti, tra cui endocrinologi, chirurghi, pediatri, ortopedici e neuropsichiatri. Vari trattamenti che coinvolgono interventi chirurgici e farmaci sono utilizzati per curare il gigantismo. Pegvisomant è un farmaco farmaceutico che ha ricevuto attenzione per essere una possibile via di trattamento per il gigantismo. La riduzione dei livelli di IGF-I a seguito della somministrazione di pegvisomant può essere incredibilmente benefica per i pazienti pediatrici con gigantismo. Dopo il trattamento con pegvisomant, gli alti tassi di crescita, una caratteristica caratteristica del gigantismo, possono essere significativamente ridotti. Pegvisomant è considerato una potente alternativa ad altri trattamenti come gli analoghi della somatostatina, un metodo di trattamento comune per l’acromegalia, se il trattamento farmacologico è associato alle radiazioni. Trovare il livello ottimale di pegvisomant è importante in modo che la normale crescita corporea non venga influenzata negativamente. A tal fine, la titolazione del farmaco può essere utilizzata per trovare il livello di somministrazione corretto.
Prognosi
Nei pazienti con gigantismo che superano l’infanzia, la prognosi è in genere buona e l’aspettativa di vita è normale.
Leggi anche:
- Quando essere troppo alti è una malattia: il gigantismo
- Differenza tra gigantismo ed acromegalia
- Acromegalia: cause, sintomi, diagnosi e trattamento
- Sindrome angio-osteoipertrofica (sindrome di Klippel-Trénaunay): cause, sintomi, diagnosi, cure
- Sindrome di Parkes Weber: cause, sintomi, diagnosi, terapie, prognosi
- Macrodistrofia lipomatosa: cause, sintomi, diagnosi, terapie
- Sindrome di Proteo: storia, cause, sintomi, diagnosi, terapie, complicanze
- Sindrome CLOVES: cause, sintomi, complicanze, diagnosi e terapia
- Sindrome di Sotos: cause, sintomi, diagnosi, terapia e prognosi
- Sindrome di Weaver-Smith: cause, sintomi, diagnosi, terapia e prognosi
- Emimegaloencefalia: cause, sintomi, diagnosi, terapia e prognosi
- Sindrome di Proteo: l’uomo che non può smettere di crescere
- Il mio mito è invece Frederick Treves
- Nanismo: sintomi, cura, cause, terapia, diagnosi e prevenzione
- Acondroplasia: diagnosi prenatale ecografica e aspettativa di vita
- Altezza: quando si può parlare di nanismo o gigantismo
- Quanto è alto l’uomo più alto del mondo?
- Sindrome di Sturge-Weber: cause, trasmissione, sintomi, diagnosi, cure
- Neurofibromatosi di tipo 1 (malattia di von Recklinghausen)
- Neurofibromatosi di tipo 2 (variante di Gardner e di Wishart)
- Sindrome di Rubinstein-Taybi: cause, sintomi, complicanze, diagnosi, terapie
- Vene varicose: sintomi iniziali e come curarle ed eliminarle
- Flebite: cos’è, quando preoccuparsi, terapie, esercizi, è mortale?
- Flebotrombosi: cause, sintomi, rischi e trattamento
- Trombosi venosa profonda: cause, terapie, tempi di guarigione, rischi
- Differenza tra flebite, flebotrombosi, tromboflebite, trombosi venosa profonda
- Tromboflebite superficiale: significato, diagnosi, cure, rischi
- Malformazioni artero-venose cerebrali: sintomi e cura
- I 12 segni che indicano una cattiva circolazione da non sottovalutare
- Formicolio alle mani, piedi, braccia e gambe: cause e cure
- Coumadin: quando si usa, dosaggio ed effetti collaterali (foglio illustrativo)
- Ho dimenticato di assumere l’anticoagulante, cosa fare?
- Cardioaspirin 100mg: effetti indesiderati, a cosa serve, dosaggi (foglio illustrativo)
- Differenza tra trombosi arteriosa e venosa profonda e superficiale
- Differenza tra aterosclerosi e arteriosclerosi
- Differenza tra trombo, embolo, coagulo, embolia e trombosi
- Differenza tra arterie, vene, capillari, arteriole e venule
- Differenza tra arteriola afferente ed efferente: struttura e funzioni
- Differenza tra circolazione sistemica, polmonare ed intracardiaca
- Segno di Collier: la retrazione della palpebra superiore dell’occhio
- Catetere venoso periferico: posizionamento e gestione infermieristica
- Cos’è la pressione venosa centrale e perché si misura?
- Differenza tra pressione venosa centrale e periferica
- Qual è la differenza tra arteria e vena?
- Trombo: cause, classificazione, trombosi venose, arteriose e sistemiche
- Differenza tra trombo e placca aterosclerotica
- Fenomeno di Raynaud: cause, sintomi e trattamento
- Glomerulo renale: schema, funzione e flusso ematico renale
- Filtrazione glomerulare, riassorbimento e secrezione
- Com’è fatto il cuore, a che serve e come funziona?
- Differenza tra pressione arteriosa e venosa
- Differenza tra pressione massima (sistolica), minima (diastolica) e differenziale
- Pressione arteriosa: valori normali e patologici
- Autoregolazione ed importanza del flusso sanguigno cerebrale
- Pressione alta (ipertensione arteriosa): sintomi, cause, valori e cure
- Perché la pressione arteriosa alta (ipertensione) è pericolosa?
- Ipertensione: cibi consigliati e da evitare per abbassare la pressione sanguigna
- Fattori di rischio cardiovascolare modificabili e non modificabili
- Differenza tra pressoterapia e linfodrenaggio
- E’ meglio il massaggio linfodrenante manuale o la pressoterapia?
- Differenza tra massaggio drenante e massaggio linfodrenante
- Differenza tra pressoterapia e cavitazione: quale preferire?
- Differenza tra pressoterapia e mesoterapia: quale preferire?
- Differenza tra massaggio linfodrenante e pressoterapia
- Linfodrenaggio manuale con metodo Vodder e Leduc: controindicazioni e tecniche
- Differenza tra pressoterapia e radiofrequenza: quale preferire?
- Effetto Doppler: cos’è e come viene usato in campo medico?
- Ecocolordoppler: cos’è, a che serve e come funziona?
- Ecocolordoppler arterioso e venoso degli arti inferiori e superiori
- Ecocolordoppler dei tronchi sovraortici: come si effettua e quali patologie studia
- Differenza tra grande safena e piccola safena
- Differenza tra arteria femorale, poplitea, tibiale anteriore e del piede
- Differenza tra arteria brachiale, radiale, ulnare e della mano
- Differenza tra vena ascellare, brachiale, radiali, ulnari, cefalica e basilica
- Differenza tra vena grande safena, femorale e poplitea dell’arto inferiore
- Cos’è la pressione sistemica di riempimento?
- Emogasanalisi arterioso: procedura, interpretazione, è dolorosa?
- Differenza tra emogasanalisi arterioso e venoso
- Saturazione dell’ossigeno: valori normali e patologici in anziani e bambini
- Perché la pressione venosa è inferiore a quella arteriosa
- Come si misura la pressione arteriosa? Guida facile
- Coagulazione intravascolare disseminata (CID): cause e terapie
- Coagulazione del sangue, cascata coagulativa, fibrinolisi: spiegazione e schema
- Test della coagulazione PT, INR PTT, aPTT, TT: valori e significato
- Tempo di protrombina (PT): valori normali, INR alto, basso, cosa fare
- Tempo di tromboplastina parziale (PTT, aPTT): alto, basso, conseguenze
- Tempo di trombina (TT): valori normali, alto, basso, significato
- D-dimero alto in tumori, mestruazioni, gravidanza, infezioni
- Fibrina e fibrinogeno: valori, alto, basso e significato
- Emofilia: cos’è, diagnosi, sintomi, tipi, terapia e cura
- Emostasi fisiologica e chirurgica: significato e fasi
- Emocromo: guida completa a tutti i valori del sangue normali e patologici
- Ematocrito (HCT): basso, alto, in gravidanza, valori normali e interpretazione
- Emocromo: valori di riferimento e significato clinico [SCHEMA]
- Triade di Virchow: i tre fattori di rischio per la trombosi
- Policitemia: sintomi, falsa, valori, cura, primaria e secondaria
Dott. Emilio Alessio Loiacono
Medico Chirurgo
Direttore dello Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o unisciti al nostro gruppo Facebook o ancora seguici su Twitter, su Instagram, su YouTube, su LinkedIn, su Tumblr e su Pinterest, grazie!
