
 Con il termine “facomatosi” (in inglese “phakomatoses”) o con “ectodermosi congenite”, in medicina si identifica un gruppo di malattie ereditarie che interessano la cute e altri organi, tra cui il cervello. In tale gruppo possiamo includere varie patologie, tra cui:
Con il termine “facomatosi” (in inglese “phakomatoses”) o con “ectodermosi congenite”, in medicina si identifica un gruppo di malattie ereditarie che interessano la cute e altri organi, tra cui il cervello. In tale gruppo possiamo includere varie patologie, tra cui:
- neurofibromatosi;
- sclerosi tuberosa;
- angiomatosi retino-cerebellare (malattia di Von Hippel-Lindau);
- angiomatosi cutanea associata ad anomalie del SNC (angiomatosi meningofacciale con calcificazione cerebrale o sindrome di Sturge-Weber);
- incontinentia pigmenti;
- sindrome di Bonnet – Dechaume – Blanc (sindrome di Wyburn-Mason);
- atassia-teleangectasia (sindrome di Louis-Bar).
La neurotibromatosi e la sclerosi tuberosa, due facomatosi tra le più diffuse, sono caratterizzate dalla presenza, nel sistema nervoso centrale (SNC), di formazioni benigne similtumorali (amartomi) che hanno la potenzialità di subire una trasformazione neoplastica. In questo articolo ci occuperemo della neurofibromatosi di tipo 1 (malattia di von Recklinghausen).
Neurofibromatosi
La neurofibromatosi è una malattia ereditaria che colpisce le cellule nervose e muco-cutanee dovuta a turbe dell’istogenesi. È caratterizzata dalla presenza di numerosi tumori benigni e maligni fibrosi (fibromi) della pelle e del tessuto nervoso (neurofibromi). Si distinguono 2 tipi, il tipo 1, detto anche malattia di von Recklinghausen, dal patologo tedesco von Recklinghausen che la descrisse nel 1882, e il tipo 2 che è molto più raro e colpisce anche i nervi ottici, uditivi e il cervello. Le due forme sono caratterizzate da ereditarietà genetica autosomica dominante; questo vuol dire che basta avere una copia di un gene mutato per avere la malattia. I figli nati dall’incrocio fra un genitore eterozigote malato e uno omozigote sano hanno il 50% di possibilità di avere anche loro la malattia. NF1 è 10 volte più comune della NF2. Interessa oltre il 90% dei casi e colpisce un nato ogni 3500-4000 persone circa: si calcolano oltre un milione e mezzo di malati nel mondo, di cui almeno 20.000 in Italia. Nonostante, come indicato dal nome, nella neurofibromatosi siano presenti neurofibromi (in numero sempre maggiore di uno), questi non ne costituiscono la caratteristica peculiare; infatti la diagnosi della malattia richiede la presenza concomitante di altre caratteristiche specifiche e riconoscibili. In assenza di tali sintomi la presenza di un neurofibroma semplice va considerata come una manifestazione clinica isolata senza alcun rapporto con la neurofibromatosi.
Neurofibromatosi di tipo 1
La neurofibromatosi di tipo 1 (NF1) è una malattia genetica autosomica dominante che colpisce una persona su circa 3500-4000 e spesso non è diagnosticata poiché provoca solo problemi estetici.
Epidemiologia
In questa malattia ereditaria la cute, il sistema nervoso, le ossa, le ghiandole endocrine e talvolta altri organi sono sede di masse similtumorali a potenziale di crescita limitato (cioè amartomi). Quelle della cute e dei nervi sono in genere schwannomi. Nell’iride i piccoli amartomi sono chiamati noduli di Lisch. La prevalenza della malattia è di 40/100000 soggetti, oppure circa un caso ogni 2500-3000 nascite. L’ereditarietà è autosomica dominante.
Sintomi e segni
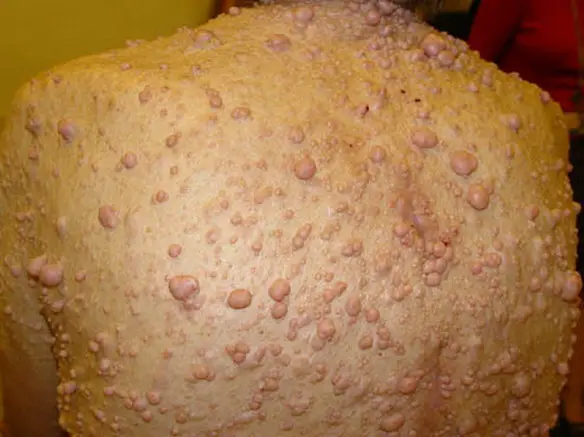
Sono caratteristiche le chiazze di iperpigmentazione (color caffèlatte) e i tumori multipli cutanei e sottocutanei, che aumentano di numero durante la seconda infanzia e l’adolescenza. Schwannomi e neurofibromi possono formarsi sulle radici spinali e sui nervi cranici, alcuni in posizione tale da comprimere più radici nervose e il midollo spinale. Spesso tali lesioni rimangono asintomatiche per un lungo periodo; a volte si aggiunge alla sindrome la presenza di meningiomi. Un’altra complicanza grave nella NF di tipo I, è la presenza di un amartoma o di un glioma a livello di uno o di entrambi i nervi ottici. Alcuni dei tumori cutanei, invece di sporgere in superficie come papillornì, ispessiscono la cute in modo diffuso (neurorna plessiforme) e sfigurano il volto come in questo caso o altre parti del corpo. In circa il 2-5% dei casi uno o più neurofibromi vanno incontro a degenerazione maligna.
Diagnosi
La diagnosi può esser sospettata sin dai primi anni di vita per la presenza sulla pelle di almeno 6 macchie color caffellatte. Fra le tecniche diagnostiche ancora sperimentali è stata utilizzata la retinografia ad infrarossi. Con il passare del tempo compaiono i neurofibromi, che si manifestano come tumori cutanei nodulari, rossastri, multipli, di consistenza soffice e localizzati in varie parti del corpo. Ciascun segno però non è sufficiente per una diagnosi di NF1; come segni minimi richiesti per avanzare una diagnosi di NF1 l’individuo deve avere almeno due di questi sintomi seguenti:
- Sei o più macchie dermatologiche marrone chiaro (le cosiddette “macchie caffellatte”), che devono essere di diametro maggiore di 5 mm prima della pubertà e di diametro maggiore di 15 mm dopo la pubertà.
- Almeno due neurofibromi.
- Almeno due escrescenze sull’iride dell’occhio.
- Crescita anormale della colonna vertebrale (scoliosi).
Nel 20% dei casi si associano alcune gravi manifestazioni (tumori cerebrali ed extracerebrali – in particolare del nervo ottico, del surrene, delle ossa – scoliosi, convulsioni, ritardo mentale, ipertensione, bassa statura, pubertà anticipata, ecc.), mentre il 30-40% dei soggetti presenta dei disturbi di apprendimento (scarsa memoria o concentrazione, difficoltà di lettura, di scrittura o di linguaggio) che necessitano di un supporto adeguato sin dalla prima infanzia. Una complicanza della NF1 è anche la pseudoartrosi congenita della tibia. Altro aspetto caratteristico è inoltre l’associazione con amartomi dell’iride, i cosiddetti noduli di Lisch.
Terapia e prognosi
Il trattamento dei tumori periferici, dei meningiomi e dei gliomi consiste nell’asportazione chirurgica (se possibile) o nella terapia radiante. L’imprevedibile decorso della malattia e la mancanza di una cura risolutiva impongono di effettuare periodici controlli medici multispecialistici presso sedi qualificate, grazie ai quali poter eventualmente intervenire con cure sintomatiche, tanto più efficaci se precoci. In entrambi i casi è quasi sempre possibile intervenire asportando i fibromi e praticando cure a base di chemioterapia e radioterapia; tuttavia i fibromi spesso ricrescono dopo pochi mesi. La struttura del cervello è normale, ma persistono stipiti cellulari anomali che continuano a differenziarsi.
Per approfondire: Neurofibromatosi di tipo 2 (variante di Gardner e di Wishart)
Leggi anche:
- Visita neurologica: svolgimento, esami, patologie, quando è necessaria?
- Sfigurata dalla neurofibromatosi, dopo il trapianto di faccia ha un volto nuovo
- Sclerosi multipla: cause, sintomi, diagnosi e prognosi
- Sclerosi laterale amiotrofica (SLA): cause, sintomi, diagnosi e prognosi
- Test di Romberg: cos’è, a che serve, come si esegue
- Differenze tra sclerosi laterale amiotrofica e sclerosi multipla
- Atrofia muscolare progressiva: cause, sintomi, cura, aspettativa di vita
- Atrofia muscolare spinale: sintomi, trasmissione, tipi e cure
- Differenze tra atrofia muscolare progressiva e sclerosi laterale amiotrofica
- Riflesso di Babinski positivo: sintomi, diagnosi, come evocarlo
- Segno di Babinski positivo nel neonato e nel bambino: che significa?
- Segno di Babinski nella sclerosi multipla e nella SLA
- Segno di Brudzinski positivo e negativo: semeiotica nella meningite
- Segno di Hoffman positivo in SLA e sclerosi multipla
- Segno di Kernig positivo e negativo: semeiotica nella meningite
- Segno di Lasègue positivo e negativo in semeiotica
- Segno di Wasserman (Lasègue inverso) positivo in semeiotica
- Segno di Binda: cos’è e come si esegue
- Segno di Amoss (o del tripode): cos’è e come si esegue
- Segno di Magnus-De Klein: cos’è e come si esegue
- Malattia di Huntington: cos’è, ereditarietà, come si trasmette, età di insorgenza
- Differenza tra tremori, spasmi, miotonia, crampi, fascicolazioni, tic
- Differenza tra discinesia, ipocinesia, ipercinesia, tardiva e primaria
- Fascicolazioni muscolari, il tremolio spontaneo di un muscolo: cause e cure
- Differenza tra astenia, ipostenia, miastenia, ipotonia, nevrastenia, iperstenia, ipertonia
- Astenia, quando mancano le forze fisiche o mentali: cause, diagnosi, cure
- Nevrastenia (esaurimento nervoso): cause, diagnosi, cure
- Ipostenia (miastenia): cause, sintomi, diagnosi, terapie, complicanze, prognosi
- Ipotonia e atonia: definizione, etimologia, significato, esempi
- Ipotonia muscolare in neonati, adulti, anziani: cause, sintomi, cure, consigli
- Ipotonia nel lattante: cause, sintomi, diagnosi e trattamento
- Iperstenia: definizione, significato, etimologia, fisiologica e patologica
- Ipertonia: definizione, significato, etimologia
- Ipertonia muscolare: cause, tipi, sintomi, diagnosi, terapie
- Cos’è il tono muscolare, a che serve, come mantenerlo?
- Distonia focale e generalizzata: tipi, sintomi, diagnosi e terapie
- Miotonia: cause, sintomi, diagnosi e terapie
- Demenza senile: cause, sintomi, decorso e cure
- Demenza da corpi di Lewy: cause, decorso, Parkinson, aspettativa di vita
- Differenza tra morbo di Alzheimer, demenza senile, vascolare e reversibile
- Mielite: infettiva, cervicale, dorsale, trasversa, si guarisce?
- Meningite: contagio, sintomi, vaccino, gravità e profilassi
- Segni meningei e irritazione meningi in bambini ed adulti
- Meningite: incubazione, fulminante, sintomi, contagio e cura
- Encefalite: conseguenze, è contagiosa, danni, si guarisce?
- Encefalite autoimmune da anticorpi anti-NMDA: trattamento, sintomi
- Meningismo: triade, segni, cause, diagnosi, definizione e cura
- Differenza tra meningite e meningismo: qual è più grave?
- Differenza tra meningite, encefalite, meningoencefalite, encefalomielite
- Differenza tra meningite virale e batterica
- Differenza tra virus e batteri: chi è più pericoloso? Diagnosi, sintomi e terapia
- Puntura lombare: complicanze, risultati, è dolorosa, a che serve?
- Meningi: anatomia, funzioni e patologia in sintesi
- Liquido cefalorachidiano: dove si trova, perdita dal naso, prelievo
- Mielopatia: significato, tipi, sintomi, diagnosi,cure, consigli, prognosi
- Segni meningei e irritazione meningi in bambini ed adulti
- Tumore al cervello: cause, sintomi iniziali e tardivi, diagnosi, cura, sopravvivenza, aspettativa di vita
- Ipertensione endocranica: valori, cause, bradicardia, terapie
- Pseudotumor cerebri (ipertensione endocranica benigna) cause e cure
- Operato al cervello per un tumore mentre suona il clarinetto
- Tumore al cervello: operato mentre suona la chitarra e canta Yesterday
- Esoftalmo bi- e monolaterale: cause, gravità, conseguenze e cure
- Transilluminazione della testa di un neonato per la diagnosi di idrocefalo
- Idrocefalo: cause, terapia, conseguenze, aspettativa di vita
- Idrocefalo nel feto e neonatale: conseguenze e cura
- Pressione intracranica e pressione di perfusione cerebrale
- Sindrome della sella vuota: cause, sintomi, diagnosi e terapie
- Differenza idrocefalo iperteso, normoteso, comunicante, ostruttivo
- Macrocefalo in neonato e bambino: sintomi, cure e ritardo psicomotorio
- Shunt cerebrale e intervento per il drenaggio permanente
- Ventricoli cerebrali: anatomia e funzioni in sintesi
- Emorragia cerebrale da caduta e trauma cranico: sintomi, diagnosi e cure
- Emorragia cerebrale: non operabile, coma, morte, si può guarire?
- Emorragia cerebrale: operazione e tempi di riassorbimento
- Emorragia cerebrale: conseguenze, riabilitazione e recupero
- Emorragia subaracnoidea: cause, conseguenze, linee guida
- Ematoma subdurale: cos’è, da quale malattia è provocata, recidivo, decorso
- Coma da emorragia cerebrale: quanto può durare?
- Differenza tra proencefalo, mesencefalo, romboencefalo, telencefalo, diencefalo
- Differenza tra metencefalo e mielencefalo
- Emicrania con aura: cause, sintomi, diagnosi e trattamenti
- Emicrania senza aura: cause, sintomi, diagnosi e trattamenti
- Differenza tra emicrania con aura ed emicrania senza aura
- Cos’è l’aura emicranica?
- Ictus cerebrale emorragico e ischemico: cause, sintomi, diagnosi, cure, rischi
- Differenza tra ictus cerebrale ed attacco ischemico transitorio (TIA)
- Che cos’è un attacco ischemico transitorio (TIA)? Impara a riconoscerlo e potrai salvare una vita, anche la tua
- Aneurisma cerebrale rotto e non rotto: cause, sintomi, diagnosi e cura
- Cosa si prova e cosa succede quando si rompe un aneurisma cerebrale?
- Ictus, emorragia cerebrale cerebrale e TIA: cosa fare e cosa assolutamente NON fare
- Sindrome frontale o ipofrontalità: cause, sintomi, conseguenze, diagnosi, cure
- Malformazioni artero-venose cerebrali: sintomi e cura
- Ischemia: cos’è, cause, conseguenze, rischi, cure
- Necrosi: significato, definizione, sinonimo, cause, cure
- Trombo: cause, classificazione, trombosi venose, arteriose e sistemiche
- Infarto, ischemia, necrosi, aterosclerosi, trombo, embolo, ictus, miocardio… Facciamo chiarezza
- Tronco cerebrale (mesencefalo, ponte e bulbo) anatomia e funzioni in sintesi
- Morbo di Parkinson: cause, sintomi, decorso, terapie
- Morbo di Alzheimer: cause, sintomi, decorso, terapie
- Parestesie: significato, cause, rischi, diagnosi, cure, rimedi, esercizi
- Epilessia infantile ed in adulti: cause, sintomi, diagnosi, cosa fare
- Epilessia: come riconoscere un attacco e soccorrere un ammalato
- Differenza tra epilessia e convulsioni
- Differenza tra epilessia e sincope
- Differenza tra epilessia parziale e generalizzata
- Epilessia: riconoscere in tempo l’arrivo di una crisi e come comportarsi
- Epilessia infantile: come comportarsi col proprio figlio?
- Esame della sensibilità tattile, dolorifica, termica, vibratoria in neurologia
- Esame delle funzioni motorie e dei riflessi in neurologia
- Esame delle funzioni cerebrali superiori (corticali) in neurologia
- Asterissi (asterixis) in neurologia: caratteristiche, significato, esecuzione
- Torcicollo spasmodico e spasmi linguali, facciali, oromandibolari, della mano (distonie focali)
- Tic, ritmie, movimenti stereotipati, acatisia e trasalimento nel paziente neurologico
- Disturbi della stazione eretta e della deambulazione in neurologia
- Esame della motilità oculare e disturbi dei movimenti coniugati in neurologia
- Sordità, esame dell’udito e tecniche audiologiche speciali in neurologia
- Aprassia ideazionale, ideomotoria e melocinetica: sintomi, diagnosi, cure
- Aprassia: significato, etimologia, tipi, zone del cervello coinvolte
- Aprassia: test usati per la diagnosi e tipici errori aprassici
- Aprassia costruttiva (apractognosia): cause, sintomi, diagnosi, terapie
- Disartria: cause, sintomi, diagnosi, trattamento
- Perdita della coordinazione muscolare: l’atassia
- Differenze tra aprassia, atassia, disprassia, afasia, disartria, anartria
- Non riconoscere i volti dei propri cari: la prosopagnosia, cause, test e cure
- Spina bifida e difetti di chiusura del tubo neurale nel feto: trasmissione, prevenzione, diagnosi e cura
- Mielomeningocele, spina bifida e schisi vertebrale: complicanze, cura, prevenzione
- Encefalocele: cause, sintomi, diagnosi, cura e prevenzione
- Anche gli esseri umani possono avere una coda
- Sindrome di Arnold-Chiari: linguaggio, aspettative di vita, invalidità, mortalità
- Siringomielia (malattia di Morvan): cause, sintomi, diagnosi, cure
- Siringobulbia: cause, sintomi, diagnosi e trattamento
- Glasgow Coma Scale per la classificazione del coma
- Pediatric Glasgow Coma Scale in italiano: scala pediatrica del coma
- Differenza tra toracentesi, paracentesi e rachicentesi
- Narcolessia: cause, sintomi, cure e terapia farmacologica
- Catatonia: significato, definizione, cause, sinonimi e cure
- Cataplessia: causa, significato, nel sonno, cura ed etimologia
- Catalessia in medicina: cause, sintomi, nel sonno e cure
- Differenza tra catatonia, catalessia e cataplessia
- Paralisi del sonno e allucinazioni ipnagogiche: cause, pericoli, rimedi
- Morte cerebrale: diagnosi, sintomi, risveglio, durata, si può guarire?
- Coma: cause, risveglio, tipi, quanto dura, fasi, segni, irreversibile
- Differenza tra coma e coma farmacologico
- Elettroencefalogramma: preparazione, alterazioni, costo, rischi
- Sindrome locked-in: cause, riabilitazione, respirazione, cure
- Stato di minima coscienza: evoluzione, risveglio, riabilitazione
- Stato vegetativo: risveglio, riabilitazione, durata e caratteristiche
- Differenza tra stato vegetativo, di minima coscienza, coma, sonno e stato soporoso
- Differenza tra sindrome locked-in e stato vegetativo
- Differenza tra stato vegetativo persistente e permanente
- Differenza tra stato vegetativo e stato di minima coscienza
- Commozione cerebrale: cos’è, cosa fare, conseguenze, tempi di recupero
- Differenza tra commozione cerebrale, trauma cranico e contusione cerebrale
- Trauma cranico: ematoma, commotivo, sintomi tardivi, cosa fare
- Emorragia cerebrale: cause, sintomi premonitori, diagnosi e cura
- Differenza tra morte clinica, biologica, legale, apparente, improvvisa ed istantanea
- Poligono di Willis: anatomia e varianti anatomiche
- Emiplegia destra, sinistra, spastica, flaccida: significato e riabilitazione
- Emiparesi destra, sinistra, facciale e neonatale: cause, sintomi e cure
- Paraplegia: etimologia, significato, sintomi, cura e riabilitazione
- Tetraplegia: significato, cause, cure e riabilitazione
- Diplegia: definizione, cause e sintomi
- Differenza tra emiplegia, emiparesi, diplegia, paraplegia, tetraplegia
- Differenza emiparesi, diparesi, tetraparesi, monoparesi, triparesi
- Differenza tra paraplegia e diplegia
- Classificazione generale delle paresi e delle plegie
- Anosognosia e Sindrome neglect: significato, test e trattamento
- Sindrome neglect (negligenza spaziale unilaterale): cura e riabilitazione
- Sistema nervoso: com’è fatto, a che serve e come funziona
- Sistema nervoso simpatico: funzioni
- Sistema nervoso parasimpatico: funzioni
- Differenza tra afasia, disartria ed aprassia
- Area di Broca: funzioni ed afasia di Broca
- Area di Wernicke: funzioni ed afasia di Wernicke
- Differenza tra afasia di Broca e di Wernicke
- Cervelletto: anatomia esterna ed interna
- Cervelletto: le lesioni cerebellari più comuni
- Le funzioni del cervelletto: apprendimento e correzione dei movimenti del corpo
- Com’è fatto il cervello, a che serve e come funziona la memoria?
- Cervello maschile e femminile: quali sono le differenze?
- Sistema nervoso autonomo simpatico e parasimpatico: anatomia e funzioni
- Ipotalamo: anatomia, struttura e funzioni
- Differenze tra ipotalamo, ipofisi, neuroipofisi e adenoipofisi
- Differenza tra midollo osseo e spinale
- Differenza tra sistema nervoso centrale e periferico: anatomia e funzioni in sintesi
- A cosa serve il midollo osseo?
- Differenza tra midollo osseo e cellule staminali
- Differenza tra midollo spinale e allungato
- Differenza tra epifisi, diafisi, metafisi ed ipofisi
- Patologie di ipotalamo e ipofisi
- Ipofisi (ghiandola pituitaria): anatomia, funzioni e ormoni secreti
- Asse ipotalamo-ipofisario: fisiologia e ormoni rilasciati
- Si può morire di epilessia?
- Sindrome pseudobulbare: cause, sintomi, diagnosi e terapie
- Malattia di Binswanger: cause, sintomi, diagnosi, cure
- Sistema piramidale e fascio genicolato: anatomia, decorso, funzioni
- Sistema extrapiramidale: anatomia, decorso, vie, funzioni, patologie
- Differenza tra via piramidale ed extrapiramidale
- Miclono: cause, sintomi, caratteristiche, quando preoccuparsi, cure
- Spasmi muscolari e mioclonie: da cosa sono causati?
- Spasmi muscolari e mioclonie: cause, diagnosi e cura delle contrazioni involontarie
- Spasmi muscolari e mioclonie: cura, trattamento e rimedi
- Spasmi muscolari e mioclonie: come si fa la diagnosi?
Dott. Emilio Alessio Loiacono
Medico Chirurgo
Direttore dello Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o unisciti al nostro gruppo Facebook o ancora seguici su Twitter, su Instagram, su YouTube, su LinkedIn, su Tumblr e su Pinterest, grazie!
