
 La malattia di Creutzfeldt-Jakob (da cui l’acronimo “MCJ“, anche chiamata “encefalopatia spongiforme subacuta” o “disturbo neurocognitivo da malattia da prioni“, un tempo nota come “morbo di Creutzfeldt-Jakob“; in inglese “Creutzfeldt–Jakob disease” da cui l’acronimo “CJD“, anche chiamata “subacute spongiform encephalopathy” o “neurocognitive disorder due to prion disease“), è una malattia cerebrale degenerativa inevitabilmente fatale, per cui attualmente non esistono cure. La malattia di Creutzfeldt-Jakob è classificata come un tipo di encefalopatia spongiforme trasmissibile (da cui l’acronimo “EST“; in inglese “transmissible spongiform encephalopathy” da cui l’acronimo “TSE“). Le encefalopatie spongiformi trasmissibili sono malattie neurodegenerative anche chiamate “malattie da prioni” ed includono, tra le altre, anche la malattia di Gerstmann-Sträussler-Scheinker (GSS), l’insonnia familiare fatale (IFF), la kuru, l’encefalopatia spongiforme bovina (BSE, comunemente conosciuta come morbo della mucca pazza) nei bovini, la malattia del deperimento cronico del cervo (CWD), la scrapie nelle pecore e la malattia di Alpers dei neonati. Attualmente non esiste un trattamento specifico per la malattia di Creutzfeldt-Jakob. Gli oppioidi possono essere usati per alleviare il dolore, mentre il clonazepam o il valproato di sodio possono aiutare con i movimenti involontari. La patologia è mortale in pochi mesi o anni nel 100% dei casi.
La malattia di Creutzfeldt-Jakob (da cui l’acronimo “MCJ“, anche chiamata “encefalopatia spongiforme subacuta” o “disturbo neurocognitivo da malattia da prioni“, un tempo nota come “morbo di Creutzfeldt-Jakob“; in inglese “Creutzfeldt–Jakob disease” da cui l’acronimo “CJD“, anche chiamata “subacute spongiform encephalopathy” o “neurocognitive disorder due to prion disease“), è una malattia cerebrale degenerativa inevitabilmente fatale, per cui attualmente non esistono cure. La malattia di Creutzfeldt-Jakob è classificata come un tipo di encefalopatia spongiforme trasmissibile (da cui l’acronimo “EST“; in inglese “transmissible spongiform encephalopathy” da cui l’acronimo “TSE“). Le encefalopatie spongiformi trasmissibili sono malattie neurodegenerative anche chiamate “malattie da prioni” ed includono, tra le altre, anche la malattia di Gerstmann-Sträussler-Scheinker (GSS), l’insonnia familiare fatale (IFF), la kuru, l’encefalopatia spongiforme bovina (BSE, comunemente conosciuta come morbo della mucca pazza) nei bovini, la malattia del deperimento cronico del cervo (CWD), la scrapie nelle pecore e la malattia di Alpers dei neonati. Attualmente non esiste un trattamento specifico per la malattia di Creutzfeldt-Jakob. Gli oppioidi possono essere usati per alleviare il dolore, mentre il clonazepam o il valproato di sodio possono aiutare con i movimenti involontari. La patologia è mortale in pochi mesi o anni nel 100% dei casi.
Eponimo e cenni storici
La denominazione “malattia di Creutzfeldt-Jakob” fu introdotta da Walther Spielmeyer nel 1922, in seguito agli studi dei neurologi tedeschi Hans Gerhard Creutzfeldt e Alfons Maria Jakob, che la descrissero per la prima volta nel 1920. Alcuni dei criteri descritti nei primi lavori relativi alla malattia, non corrispondono ai criteri attuali ed è stato ipotizzato che almeno due dei pazienti osservati negli studi iniziali e descritti come sofferenti della malattia di Creutzfeldt-Jakob, siano in realtà stati affetti da una malattia diversa. Una prima descrizione della forma familiare si ha dal neurologo e psichiatra tedesco Friedrich Meggendorfer (1880-1953). La nuova variante della malattia di Creutzfeldt-Jakob (vMCJ) è stata dentificata per la prima volta nel 1996. Nel 1997 a Stanley B. Prusiner dell’Università della California, San Francisco (UCSF) è stato assegnato il Premio Nobel per la fisiologia e per la medicina nel 1997 per la scoperta dei prioni. Nel 2004 un rapporto pubblicato su The Lancet ha dimostrato che la nuova variante della malattia di Creutzfeldt-Jakob può essere trasmessa attraverso trasfusioni di sangue. Non esistendo un test per determinare se un donatore di sangue è stato infettato con la nuova variante della malattia, il governo britannico ha impedito a chiunque avesse ricevuto una trasfusione di sangue, a partire dal gennaio 1980, la donazione di sangue. A partire dal 1999 è stato posto il divieto in Gran Bretagna per l’utilizzo del sangue del Regno Unito per la realizzazione di prodotti frazionari come l’albumina. In Germania e in Italia chi ha passato sei mesi o più di vita nel Regno Unito tra gennaio 1980 e dicembre 1996 viene permanentemente escluso dalla donazione di sangue.
Epidemiologia
La malattia di Creutzfeldt-Jakob colpisce circa una persona per milione di persone all’anno. L’esordio è tipicamente tra i 60 ed i 70 anni, tranne la nuova variante della malattia di Creutzfeldt-Jakob, che colpisce in genere tra i 25 ed i 30 anni.
Classificazione
La malattia di Creutzfeldt-Jakob è distinta in quattro forme:
- malattia di Creutzfeldt-Jakob forma sporadica (acronimo italiano: sMCJ; acronimo inglese sCJD): causata dal misfolding spontaneo della proteina prionica in un individuo. Questa forma rappresenta circa l’85% dei casi di CJD;
- malattia di Creutzfeldt-Jakob forma familiare (acronimo italiano: fMCJ; acronimo inglese fCJD): causata dalla mutazione del gene per PrP, che codifica per la proteina prionica (PRNP). Questa forma rappresenta tra l’8 ed il 15% dei casi di CJD;
- malattia di Creutzfeldt-Jakob forma acquisita iatrogena (acronimo italiano: iMCJ; acronimo inglese iCJD): è causata dalla contaminazione del paziente con il tessuto di una persona infetta, di solito come risultato di una procedura medica (CJD iatrogena). Le procedure mediche associate alla diffusione di questa forma di CJD includono la trasfusione di sangue dalla persona infetta, l’uso di ormoni della crescita ipofisaria di origine umana, la terapia con ormoni gonadotropinici e trapianti di cornea e meninge. Questa forma rappresenta circa il 5% dei casi di CJD;
- nuova variante della malattia di Creutzfeldt-Jakob (acronimo italiano: nvMCJ; acronimo inglese vCJD da “variant CJD”): forse correlata al consumo di carne bovina proveniente da animali affetti da encefalopatia spongiforme bovina o trasmessa da uomo a uomo attraverso trasfusioni di sangue.
Cause
La CJD è un tipo di encefalopatia spongiforme trasmissibile (TSE), causata da prioni. I prioni sono proteine mal ripiegate che si riscontrano nei neuroni del sistema nervoso centrale (SNC); si pensa che o prioni influenzino i processi di segnalazione, danneggiando i neuroni e provocando la degenerazione che causa l’aspetto spongiforme nel cervello colpito. Il prione CJD è pericoloso non solo in sé, ma anche perché può causare il mal ripiegamento anche delle proteine normalmente ripiegate. Il numero di molecole proteiche mal ripiegate aumenterà quindi in modo esponenziale e il processo porta a una grande quantità di proteine insolubili nelle cellule colpite. Questa massa di proteine mal ripiegate interrompe la funzione delle cellule neuronali e provoca la loro morte.
Circa l’85% dei casi di malattia di Creutzfeldt-Jakob si verifica sporadicamente per ragioni sconosciute (malattia di Creutzfeldt-Jakob sporadica), mentre circa il 8-15% dei casi la malattia è familiare, ereditata con modalità autosomica dominante. Le persone possono sviluppare CJD a causa della mutazione del gene PrP, che codifica per la proteina prionica (PRNP). In casi sporadici, l’errato ripiegamento della proteina prionica è un processo che si ipotizza si verifichi come conseguenza degli effetti dell’invecchiamento sul meccanismo cellulare, il che spiega perché la malattia compare spesso più tardi nella vita. Uno studio dell’UE ha stabilito che l’87% dei casi era sporadico, l’8% genetico, il 5% iatrogeno e meno dell’1% variante.
La proteina difettosa può essere anche trasmessa da innesti corneali, innesti durali o tramite impianti di elettrodi (forma iatrogena).
La malattia inoltre è stata indicata come il risultato di un uso dell’ormone umano della crescita ottenuto dalle ghiandole pituitarie di persone che sono morte di malattia di Creutzfeldt-Jakob. Negli Stati Uniti il farmaco che utilizza l’ormone da cadavere è stato ritirato nel 1985. Si ritiene che l’uomo possa contrarre la malattia consumando carni di animali infettati con la forma bovina della malattia. Non ci sono prove che la malattia di Creutzfeldt-Jakob sporadica possa diffondersi tra le persone attraverso il normale contatto o le trasfusioni di sangue, sebbene ciò sia possibile nella variante della malattia di Creutzfeldt-Jakob. Si ritiene infine che la pratica del cannibalismo (mangiare carne umana) sia la causa di trasmissione per i prioni, causando la kuru, un tipo di encefalopatia spongiforme trasmissibile riscontrata tra le donne e i bambini della popolazione Fore in Papua Nuova Guinea.
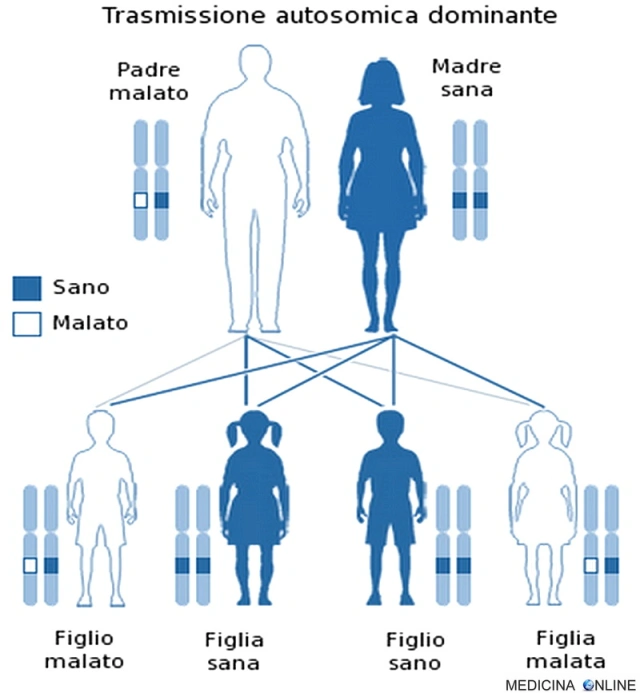
Trasmissione autosomica dominante
In circa il 8-10% dei casi la CJD è familiare, ereditata con modalità autosomica dominante. Una malattia è detta a trasmissione autosomica dominante quando basta una singola copia dell’allele difettoso per far sì che la malattia si esprima, a prescindere dal sesso (basta un solo genitore malato). Il figlio di un individuo affetto ha la probabilità del 50% di essere affetto, cioè 1 figlio su 2 è malato e può trasmettere a sua volta la malattia alla metà dei suoi figli. In questo caso non può esistere un “portatore sano” (cosa che invece si può verificare nella trasmissione autosomica recessiva): chi possiede l’allele alterato, ha la patologia, mentre chi non lo possiede è sano. Di conseguenza da due genitori sani nascono il 100% di figli sani, mentre se entrambi i genitori sono malati allora si avranno il 100% di figli malati.
Segni e sintomi
Il primo sintomo della CJD è di solito una demenza rapidamente progressiva, che porta a perdita di memoria, cambiamenti di personalità e allucinazioni. Il mioclono (movimenti a scatti) si verifica tipicamente nel 90% dei casi, ma può essere assente all’esordio iniziale. Altre caratteristiche che si verificano frequentemente includono ansia, depressione, paranoia, sintomi ossessivo-compulsivi e psicosi. Questi sintomi si associano a problemi fisici come disturbi del linguaggio, disfunzione dell’equilibrio e della coordinazione (atassia), cambiamenti nell’andatura e postura rigida. Nella maggior parte delle persone con CJD, questi sintomi sono accompagnati da movimenti involontari. Problemi di memoria, cambiamenti comportamentali, scarsa coordinazione e disturbi visivi sono in genere i segni precoci della malattia, mentre demenza, movimenti involontari, cecità, debolezza e coma sono invece sintomi e segni più tardivi.
Continua la lettura con: Malattia di Creutzfeldt-Jakob(encefalopatia spongiforme subacuta): diagnosi, cure, prognosi, mortalità
Per approfondire:
- Encefalopatie spongiformi trasmissibili (malattie da prioni): cosa sono e cosa le causa?
- Nuova variante della malattia di Creutzfeldt-Jakob: cause, sintomi, diagnosi, cure
- Encefalopatia spongiforme bovina (morbo della mucca pazza): cause, sintomi, diagnosi, cure
- Malattia di Gerstmann-Sträussler-Scheinker: cause, sintomi, diagnosi, cura, mortalità
- Prionopatia variabilmente sensibile alla proteasi: cause, sintomi, diagnosi, cure
- Malattia prionica associata a diarrea e a neuropatia autonomica: cause, sintomi, cure
- Infezioni croniche da “virus lenti” e da agenti non convenzionali (prioni)
- Insonnia fatale familiare: cause, sintomi, diagnosi, polisonnografia, cure
- Insonnia familiare fatale: diagnosi, trasmissione, cause, cure
- Disartria: cause, sintomi, diagnosi, trattamento
- Perdita della coordinazione muscolare: l’atassia
- Spasmi muscolari e mioclonie: cause, diagnosi e cura delle contrazioni involontarie
- Amiloidosi: sintomi, cura, diagnosi, prognosi, sopravvivenza
- Nistagmo: cause, sintomi, classificazione e terapia
- Corpi di Hirano: cosa sono e in quale malattie si trovano?
- Ippocampo: anatomia, funzioni e ruolo nella memoria
- Complicanze respiratorie delle malattie neuromuscolari: sintomi, segni, diagnosi
- Complicanze respiratorie delle malattie neuromuscolari: trattamento, prognosi
- Complicanze respiratorie delle malattie neuromuscolari: anatomia patologica e fisiopatologia
Leggi anche:
- Visita neurologica: svolgimento, esami, patologie, quando è necessaria?
- Riflesso di Babinski positivo: sintomi, diagnosi, come evocarlo
- Segno del ventaglio (fenomeno di Dupré); come si evoca e cosa significa?
- Segno di Bing, di Cornell, di Gonda, di Moniz, di Strümpel e altri segni simili
- Segno di Oppenheim, di Chaddock, di Gordon, di Schaeffer in medicina
- Segno di Myerson: come si evoca e cosa indica?
- Segno di Lhermitte: come si evoca e cosa significa?
- Segno di Babinski positivo nel neonato e nel bambino: che significa?
- Segno di Babinski positivo: quali patologie può indicare?
- Segno di Babinski bilaterale: cos’è e che patologia indica?
- Segno di Babinski nella sclerosi multipla e nella SLA
- Segno di Babinski ed alluce muto: cosa significa?
- Segno di Hoffman positivo in SLA e sclerosi multipla
- Segno di Brudzinski positivo e negativo: semeiotica nella meningite
- Segno di Kernig positivo e negativo: semeiotica nella meningite
- Segno di Lasègue positivo e negativo in semeiotica
- Segno di Wasserman (Lasègue inverso) positivo in semeiotica
- Segno di Binda: cos’è e come si esegue
- Segno di Amoss (o del tripode): cos’è e come si esegue
- Segno di Magnus-De Klein: cos’è e come si esegue
- Segni meningei e irritazione meningi in bambini ed adulti
- Triade di Charcot e triade neurologica di Charcot
- Pentade di Reynolds: cos’è e cosa indica?
- Malattia di Alzheimer: cause, sintomi, decorso, terapie
- Differenza tra malattia di Alzheimer, demenza senile, vascolare e reversibile
- Differenza tra malattia di Alzheimer e morbo di Parkinson: sintomi comuni e diversi
- Demenza senile: cause, sintomi, decorso e cure
- Sclerosi laterale amiotrofica (SLA): cause, sintomi, diagnosi e prognosi
- Differenze tra sclerosi laterale amiotrofica e sclerosi multipla
- Malattia di Parkinson: cause, sintomi, decorso, terapie
- Malattia di Parkinson: sintomi motori e non motori, iniziali e tardivi
- Scala di Hoehn e Yahr e aspettativa di vita nella malattia di Parkinson
- Sclerosi multipla: cause, sintomi, diagnosi e prognosi
- Sindrome di Behçet: cause, sintomi, diagnosi, trattamento
- Test di Stroop: usi, tipi, varianti e malattie indagate
- Ernia del disco L5 S1: espulsa, cure naturali, quando operare, guarigione
- Ernia del disco e mal di schiena: sintomi, diagnosi e cura
- Sindrome di Arnold-Chiari: linguaggio, aspettative di vita, invalidità, mortalità
- Siringomielia (malattia di Morvan): cause, sintomi, diagnosi, cure
- Spondilosi cervicale: cause, sintomi, cure, esercizi, invalidità
- Spondilosi lombare: cause, sintomi, terapie, esercizi, invalidità
- Idrocefalo: cause, terapia, conseguenze, aspettativa di vita
- Transilluminazione della testa di un neonato per la diagnosi di idrocefalo
- Idrocefalo nel feto e neonatale: conseguenze e cura
- Pressione intracranica e pressione di perfusione cerebrale
- Liquor: caratteristiche, alterazioni della circolazione liquorale e della pressione intracranica
- Differenza idrocefalo iperteso, normoteso, comunicante, ostruttivo
- Frattura della base cranica: cause, sintomi, diagnosi, terapie, rischi
- Differenza tra frattura di LeFort I, II e III
- Liquido cefalorachidiano: dove si trova, perdita dal naso, prelievo
- Cranio e base cranica: ossa, anatomia e funzioni
- Differenza tra frattura composta, composta, esposta e patologica
- Nervi cranici: anatomia, funzioni, schema, tabella, patologie
- Ipertensione endocranica: valori, cause, bradicardia, terapie
- Meningite batterica e virale: sintomi, profilassi, cure
- Emorragia cerebrale da caduta e trauma cranico: sintomi, diagnosi e cure
- Emotimpano: quando il sangue si raccoglie nell’orecchio
- Rinoliquorrea (rinorrea cerebrospinale): quando il liquor esce dal naso
- Otoliquorrea (otorrea cerebrospinale): quando il liquor esce dalle orecchie
- Segno di Battle (ecchimosi mastoidea): il livido dietro l’orecchio
- Segno del procione (ecchimosi periorbitale): cause e caratteristiche
- Occhio nero (ematoma periorbitale): cause e caratteristiche
- Esame della sensibilità tattile, dolorifica, termica, vibratoria in neurologia
- Esame delle funzioni motorie e dei riflessi in neurologia
- Esame delle funzioni cerebrali superiori (corticali) in neurologia
- Asterissi (asterixis) in neurologia: caratteristiche, significato, esecuzione
- Torcicollo spasmodico e spasmi linguali, facciali, oromandibolari, della mano (distonie focali)
- Tic, ritmie, movimenti stereotipati, acatisia e trasalimento nel paziente neurologico
- Disturbi della stazione eretta e della deambulazione in neurologia
- Esame della motilità oculare e disturbi dei movimenti coniugati in neurologia
- Sordità, esame dell’udito e tecniche audiologiche speciali in neurologia
- Emorragia cerebrale: non operabile, coma, morte, si può guarire?
- Emorragia cerebrale: operazione e tempi di riassorbimento
- Paraplegia: etimologia, significato, sintomi, cura e riabilitazione
- Paraplegia: erezione, disfunzione erettile ed eiaculazione
- Tetraplegia: significato, cause, cure e riabilitazione
- Differenza tra paraplegico e tetraplegico
- Atrofia muscolare spinale: sintomi, trasmissione, tipi e cure
- Sistema nervoso simpatico: funzioni
- Sistema nervoso parasimpatico: funzioni
- Sclerosi laterale amiotrofica (SLA): cause, sintomi, diagnosi e prognosi
- Atrofia muscolare progressiva: cause, sintomi, cura, aspettativa di vita
- Differenze tra atrofia muscolare progressiva e sclerosi laterale amiotrofica
- Emorragia cerebrale: cause, sintomi premonitori, diagnosi e cura
- Differenza tra ictus ischemico ed emorragico
- Che cos’è un attacco ischemico transitorio (TIA)? Impara a riconoscerlo e potrai salvare una vita, anche la tua
- Ictus, emorragia cerebrale cerebrale e TIA: cosa fare e cosa assolutamente NON fare
- Emorragia subaracnoidea: cause, sintomi, diagnosi e cura
- Malformazioni artero-venose cerebrali: sintomi e cura
- Differenza tra emorragia cerebrale e subaracnoidea
- Differenza tra emorragia cerebrale ed aneurisma
- Differenza tra emorragia cerebrale ed ictus
- Differenza tra ictus cerebrale ed attacco ischemico transitorio (TIA)
- Lesioni da decubito: prevenzione, stadi, classificazione e trattamento
- Cosa sente chi è in coma?
- Aneurisma cerebrale rotto e non rotto: cause, sintomi, diagnosi e cura
- Ragazza muore a 24 anni per un aneurisma cerebrale durante un orgasmo da masturbazione
- Tipi e grandezza degli aneurismi cerebrali
- Cosa si prova e cosa succede quando si rompe un aneurisma cerebrale?
- Tumore al cervello: cause, sintomi iniziali e tardivi, diagnosi, cura, sopravvivenza, aspettativa di vita
- Emicrania con aura: cause, sintomi, diagnosi e trattamenti
- Emicrania senza aura: cause, sintomi, diagnosi e trattamenti
- Cefalea coitale ed orgasmica: il mal di testa durante il sesso e l’orgasmo
- Diagnostica per immagini nell’aneurisma cerebrale
- Differenza tra emicrania e cefalea
- Differenza tra morte clinica, biologica, legale, apparente, improvvisa ed istantanea
- Differenza tra morte cerebrale, stato vegetativo e coma
Dott. Emilio Alessio Loiacono
Medico Chirurgo
Direttore dello Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, segui la nostra pagina Facebook o unisciti al nostro gruppo Facebook o ancora seguici su Twitter, su Instagram, su YouTube, su LinkedIn, su Reddit, su Tumblr e su Pinterest, grazie!
