
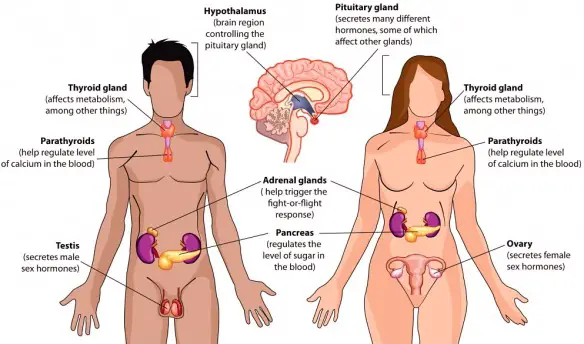
 Con sindrome da deficit polighiandolare (o sindromi polighiandolari autoimmuni, o ancora sindromi da deficit poliendocrino) si intende una sindrome caratterizzata da ipofunzione contemporanea di diverse ghiandole endocrine; solitamente sono riscontrate in più membri di una stessa famiglia.
Con sindrome da deficit polighiandolare (o sindromi polighiandolari autoimmuni, o ancora sindromi da deficit poliendocrino) si intende una sindrome caratterizzata da ipofunzione contemporanea di diverse ghiandole endocrine; solitamente sono riscontrate in più membri di una stessa famiglia.
Leggi anche: Cos’è una ghiandola endocrina? A che servono gli ormoni ed il sistema endocrino?
Cause
Il deficit endocrino può essere causato da un’infezione, un infarto o un tumore che comportino la distruzione di tutta una ghiandola endocrina o di una gran parte di essa. Tuttavia, l’attività di una ghiandola endocrina risulta il più delle volte depressa come risultato di una reazione autoimmune che determina infiammazione, infiltrazione linfocitaria e distruzione parziale o completa della ghiandola. Una malattia autoimmune che colpisce una ghiandola è frequentemente seguita dalla compromissione di altre ghiandole, dando luogo a un’insufficienza endocrina multipla.
Tipi
Sono stati descritti due quadri principali di insufficienza:
- tipo I l’esordio si verifica di solito nell’infanzia o prima dei 35 anni. L’ipoparatiroidismo è il più frequentemente riscontrato (79%), seguito dall’insufficienza corticosurrenalica (72%). L’insufficienza gonadica si verifica dopo la pubertà nel 60% delle donne e in circa il 15% degli uomini. È comune la presenza di candidosi mucocutanea cronica. Raramente è presente diabete mellito. Questo quadro può essere associato ai fenotipi HLA A3 e A28 o a un locus situato sul cromosoma 21. La trasmissione ereditaria solitamente segue una modalità autosomica recessiva;
- tipo II l’insufficienza ghiandolare si verifica generalmente negli adulti, con un picco di incidenza a 30 anni di età. Essa interessa sempre la corteccia surrenale e frequentemente la tiroide (sindrome di Schmidt) e le insule pancreatiche, producendo diabete mellito insulino-dipendente (Insulin-Dependent Diabetes Mellitus, IDDM). Sono frequentemente presenti anticorpi contro le ghiandole bersaglio, specialmente contro gli enzimi del citocromo P-450 della corteccia surrenale. Tuttavia, il loro ruolo nella patogenesi del danno ghiandolare è poco chiaro. Alcuni pazienti hanno anticorpi stimolanti la tiroide e si presentano inizialmente con segni e sintomi di ipertiroidismo. La distruzione ghiandolare è principalmente un risultato dell’autoimmunità cellulo-mediata, conseguenza di una depressione della funzione delle cellule T suppressor oppure di qualche altro tipo di danno mediato dalle cellule T. Inoltre, è comune la riduzione dell’immunità sistemica mediata dalle cellule T, rivelata da una scarsa risposta ai test cutanei con antigeni standard, come la candidina (dalla Candida), la tricofitina (dal Trichophyton) e la tubercolina. Una riduzione della reattività si dimostra anche nel 30% circa dei parenti di primo grado con funzione endocrina normale. È stato suggerito che gli specifici tipi HLA caratteristici del tipo II siano associati con una suscettibilità ad alcuni virus che inducono la reazione distruttiva.
Un gruppo ulteriore, il tipo III, si presenta negli adulti e non interessa la corteccia surrenale, ma comprende almeno due delle seguenti condizioni: deficit funzionale tiroideo, IDDM, anemia perniciosa, vitiligine e alopecia. Poiché la caratteristica distintiva del quadro di tipo III è l’assenza di insufficienza corticosurrenalica, esso potrebbe essere semplicemente un “cestino per i rifiuti” di una malattia combinata che viene convertita in tipo II se si sviluppa insufficienza del corticosurrene.
Sintomi, segni e diagnosi
La presentazione clinica dei pazienti con sindromi da deficit polighiandolare è la somma dei quadri clinici dei singoli deficit. Non esiste una sequenza specifica per la comparsa delle singole distruzioni ghiandolari. La determinazione dei livelli di anticorpi circolanti diretti contro le ghiandole endocrine o i loro componenti non sembra essere utile, poiché tali anticorpi possono persistere per anni senza che il paziente sviluppi un’insufficienza endocrina. Tuttavia, il rilievo della presenza di anticorpi è di aiuto in alcune situazioni, come per la distinzione di un iposurrenalismo su base autoimmune da un iposurrenalismo tubercolare e per la determinazione della causa di un ipotiroidismo. La presenza di deficit endocrini multipli può essere indicativa di un’insufficienza ipotalamo-ipofisaria. In quasi tutti i casi, i livelli elevati delle tropine ipofisarie dimostreranno la natura periferica del difetto; tuttavia, sono stati riportati anche rari casi di insufficienza ipotalamo-ipofisaria come parte della sindrome di tipo II.
Terapia
Il trattamento per le sindromi da deficit polighiandolare dipende dalle diverse carenze ghiandolari individuali; in genere si procede con terapia ormonale sostitutiva per correggere ciascun singolo difetto ormonale, quindi vanno trattate contemporaneamente tutte le endocrinopatie. L’interazione reciproca dei deficit multipli (ad esempio l’insufficienza corticosurrenalica combinata con il diabete mellito) può rendere complicato il trattamento. I pazienti con ipofunzione di una sola ghiandola endocrina devono essere tenuti in osservazione per anni al fine di evidenziare l’eventuale sviluppo di deficit addizionali. L’insufficienza gonadica non risponde al trattamento con ormoni gonadotropi e la candidosi mucocutanea cronica è solitamente resistente alla terapia. Se somministrate precocemente nel corso della insufficienza endocrina, dosi immunosoppressive di ciclosporina A possono essere di giovamento in alcuni pazienti.
Leggi anche:
- Diabete mellito: diffusione, sintomi, classificazione e diagnosi differenziale
- Diabete mellito: cause e fattori di rischio del tipo 1 e 2
- Diabete mellito: come si forma la malattia?
- Diabete mellito: conseguenze e complicanze a lungo termine
- Ormoni tiroidei: differenza T3 e T4, valori normali e patologici
- TSH alto, basso e valori normali: qual è il significato clinico?
- Tireoglobulina alta, bassa, valori normali ed interpretazione
- Ormoni estrogeni: cosa sono e quali funzioni svolgono?
- Progesterone: cos’è, a cosa serve, valori e quali funzioni ha in gravidanza?
- Endometriosi: cause, sintomi e menopausa
- Estrogeni, sindrome premestruale, vampate di calore e menopausa
- Aumentare gli estrogeni naturalmente e senza farmaci
- Morbo di Addison: sintomi, immagini, terapia, mortalità, aspettative di vita
- Sindrome di Cushing: immagini, sintomi, diagnosi, cura, guarigione
- Feocromocitoma e surrenectomia: sintomi e conseguenze
- Iperplasia surrenale congenita: tipi, sintomi, diagnosi, cura
- Iperplasia surrenale acquisita: cause, sintomi, terapia
- Iperandrogenismo femminile: significato, cause, sintomi e terapie
- Iperandrogenismo nell’uomo: significato, cause, sintomi e terapie
- Surrene: differenza tra ormoni della parte corticale e midollare
- Deficienza di ACTH e cortisolo (ipocortisolismo secondario)
- Deficienza di CRH e cortisolo (ipocortisolismo terziario)
- Malattia di Cushing: cause, sintomi, diagnosi, terapia
- Sindrome di Achard-Thiers
- Sindrome di Waterhouse-Friderichsen: cause e sintomi
- Iperaldosteronismo primario e secondario: tipi e sintomi
- Iperaldosteronismo primario (sindrome di Conn): cause, sintomi, terapia
- Iperaldosteronismo secondario: cause, sintomi e terapia
- Metabolismo basale: cos’è, definizione, calcolo, alto, basso, totale
- Asse ipotalamo-ipofisi-tiroide: funzionamento ed ormoni rilasciati
- Ipertiroidismo: cause, cura, valori diagnosi, sintomi iniziali, conseguenze
- Ipertiroidismo nell’uomo: sintomi, conseguenze sulla libido, cure
- Ipotiroidismo: sintomi, diagnosi, cura farmacologica e consigli dietetici
- Esame obiettivo del collo: palpazione della tiroide, video e spiegazione
- Ecografia della tiroide: a cosa serve, come si svolge e come ci si prepara all’esame
- Scintigrafia tiroidea: risultati, captazione, noduli, costo
- Agoaspirato tiroideo: esito, complicanze, dolore, referto, prezzo
- Tiroidite di Hashimoto: esami, cura, conseguenze, dieta, guarire
- Tiroidite di De Quervain (subacuta): sintomi, dieta, si guarisce, è contagiosa?
- Tireotossicosi autoimmune, iatrogena, factitia, valori, cura
- Differenza tra ipertiroidismo e tireotossicosi
- Morbo di Basedow: alimentazione, cura, occhi, si guarisce, rimedi
- Gozzo tiroideo: semplice, tossico, endemico, rimedi, intervento, immagini
- Gozzo tossico nodulare e multinodulare: sintomi, diagnosi e cura
- Alimenti gozzigeni (antitiroidei) vietati nelle alterazioni tiroidee
- Morbo di Plummer (adenoma tossico): sintomi, diagnosi e terapia
- Adenoma tiroideo: sintomi, diagnosi e trattamento
- Tiroidite acuta, cronica, autoimmune: sintomi e conseguenze
- Tiroidite post partum: sintomi, fasi, diagnosi, cura e rischi
- Iodio importante per prevenire le malattie della tiroide: in quali alimenti trovarlo?
- Sali minerali: definizione, funzioni, alimenti, integratori [GUIDA COMPLETA]
- Carenza, sovraccarico e tossicità da ferro: anemia ed emocromatosi
- Sovraccarico di ferro: emosiderosi ed emocromatosi
- Iodio: fabbisogno giornaliero, funzioni, dieta, carenza, tossicità
- Tiroidectomia totale: complicanze, postoperatorio e aumento di peso
- Tiroidectomia: cosa cambierà nella mia vita dopo l’intervento?
- Tiroidectomia: cosa mi succederà prima, durante e dopo l’intervento chirurgico
- Terapia radiometabolica dell’ipertiroidismo con radioiodio: indicazioni ed effetti collaterali
- Nodulo Tiroideo: sintomi e cure
- Eutirox: quando si usa, dosaggio ed effetti collaterali (foglio illustrativo)
- Tiroide: anatomia, funzioni e patologie in sintesi
- Eutirox per dimagrire: come si usa?
- Eutirox può essere usato anche per dimagrire? Quali sono i rischi?
- Eutirox: effetti collaterali e controindicazioni del farmaco
- Eutirox: si può assumere in gravidanza ed allattamento?
- Eutirox in sovradosaggio: cosa fare, è pericoloso, quando chiamare il medico?
- Dosaggio Eutirox: come scegliere quello corretto?
- Terapia soppressiva con Eutirox per ridurre un nodulo tiroideo
- Eutirox può causare o far aumentare l’acne?
- Come e quando assumere correttamente Eutirox: attenzione a dieta e farmaci
- Eutirox ed interazioni con altri farmaci e sostanze
- Eutirox ed effetti collaterali a lungo termine: cuore ed osteoporosi
- Tumore maligno della tiroide: carcinomi differenziati ed indifferenziati
- Carcinoma papillare della tiroide: sintomi, prognosi, mortalità
- Carcinoma follicolare della tiroide: sintomi, prognosi, mortalità
- Carcinoma midollare della tiroide: calcitonina, prognosi, mortalità
- Carcinoma anaplastico della tiroide: ecografia, diagnosi, trattamento
- Linfoma maligno tiroideo: caratteristiche, sintomi, mortalità
- Carcinoma metastatico della tiroide: sintomi, diagnosi, cura
- Neoplasie Endocrine Multiple (MEN) tipo 1 e 2
Lo Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o unisciti al nostro gruppo Facebook o ancora seguici su Twitter, su Instagram o su Pinterest, grazie!
