
La sindrome di Bannayan-Riley-Ruvalcaba (BRRS) è una malattia congenita rara con poliposi amartomatosa intestinale, lipomi, macrocefalia e lentigginosi genitale.
Cosa sono i polipi intestinali?
I polipi intestinali sono masse anomale di tessuto che si formano a partire dalla mucosa dell’intestino (specialmente nel colon e nel retto) e protrudono nel lume. Pur essendo formazioni benigne, con il tempo possono diventare maligni (carcinoma): per tale motivo motivo l’identificazione e l’eventuale asportazione dei polipi intestinali rappresenta un efficace strumento di prevenzione del cancro al colon-retto. Se il polipo aderisce direttamente alla parete dell’intestino, si definisce “polipo sessile“; se al contrario aderisce mediante un peduncolo, si definisce “polipo peduncolato“. In base alle proprie caratteristiche specifiche, i polipi possono essere di varia tipologia:
- polipi iperplastici ed infiammatori: sono di origine benigna, hanno basso rischio di trasformazione in tumore maligno e sono spesso associati a Colite ulcerosa, Morbo di Crohn, coliti infettive e diverticolosi;
- polipi amartomatosi: sono lesioni non neoplastiche frequentemente di origine familiare;
- polipi neoplastici o adenomatosi: hanno un più alto rischio di trasformazione in cancro e si distinguono in:
- polipi tubulari (a minor rischio di trasformazione neoplastica);
- polipi villosi (a maggior rischio di trasformazione neoplastica);
- polipi misti tubulo-villosi.
In base al numero, avremo:
- polipo singolo (unico): il polipo si presenta da solo;
- polipi multipli: sono presenti da 2 a 100 polipi;
- poliposi: sono presenti più di 100 polipi. La poliposi può essere:
- di origine sporadica (presentarsi da sola in un individuo la cui famiglia non soffra di poliposi);
- di origine familiare (causata da un difetto genico trasmissibile): in questo caso il rischio di trasformazione in tumore del colon-retto è elevato. Questo è proprio il caso della poliposi adenomatosa familiare, oggetto di questo articolo).
Sinonimi
Possibile sinonimo della sindrome di Bannayan-Riley-Ruvalcaba è “sindrome di Myhre-Riley-Smith”.
Epidemiologia
La prevalenza non è nota, ma è probabilmente inferiore a 1/1000000.
Cause e fattori di rischio
È causata nel 60% dei casi da mutazione nel gene omologo della fosfatasi e della tensina (PTEN; 10q23), che codifica per PTEN, una fosfatasi a doppia specificità. Se la BRRS si associa a mutazioni della linea germinale di PTEN, appartiene al gruppo delle PHTS. Dato che i geni non correlati a PTEN, ma alla CS (SDHB-D, AKT1, PIK3CA e KLLN) non sono stati formalmente esaminati nella BRRS, non è chiaro se siano coinvolti anche nella BRRS non correlata a PTEN.
Trasmissione
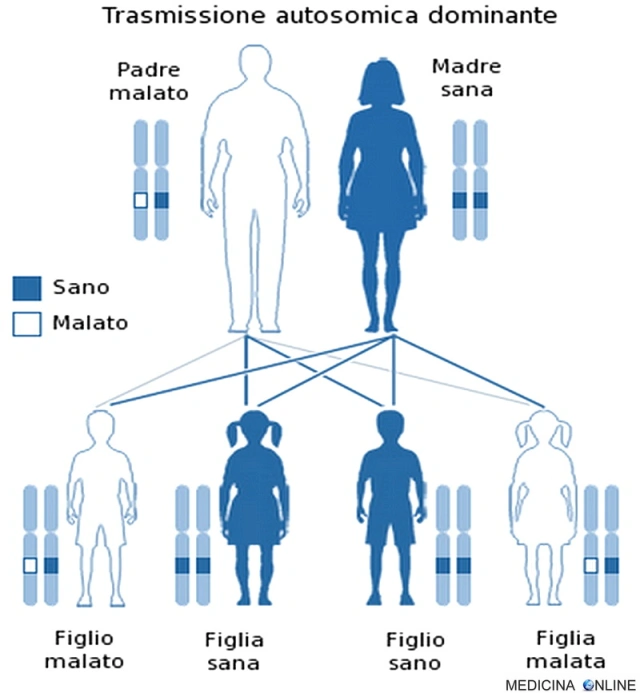
La trasmissione dell’alterazione genetica avviene per via autosomica dominante. Una malattia è detta a trasmissione autosomica dominante quando basta una singola copia dell’allele difettoso per far sì che la malattia si esprima, a prescindere dal sesso (basta un solo genitore malato). Il figlio di un individuo affetto ha la probabilità del 50% di essere affetto, cioè 1 figlio su 2 è malato e può trasmettere a sua volta la malattia alla metà dei suoi figli. In questo caso non può esistere un “portatore sano” (cosa che invece si può verificare nella trasmissione autosomica recessiva): chi possiede l’allele alterato, ha la patologia, mentre chi non lo possiede è sano. Di conseguenza da due genitori sani nascono il 100% di figli sani, mentre se entrambi i genitori sono malati allora si avranno il 100% di figli malati.
Sintomi e segni
Condivide con la sindrome di Cowden (CS) alcuni segni clinici tipici, che si manifestano con diversa frequenza. Contrariamente alla CS, il quadro classico della BRRS esordisce in epoca neonatale o immediatamente dopo con macrocefalia, struma di Hashimoto, lipomatosi, malformazioni vascolari e lentigginosi punteggiata del pene o della vulva. In un sottotipo della BRRS, si osserva ritardo dello sviluppo e poliposi amartomatosa gastrointestinale. Non è chiaro se siano tipici della BRRS alcuni segni osservati in singoli casi, come la miopatia prossimale, il pectus excavatum, l’iperestensibilità articolare, la scoliosi e l’elevato peso alla nascita. Sebbene non si ritenga che questa sindrome predisponga al cancro, i pazienti con BRRS eterozigoti per una mutazione di PTEN condividono lo stesso rischio di sviluppare il cancro dei pazienti con CS. L’età più precoce alla diagnosi dei tumori maligni della tiroide nella sindrome tumorale amartomatosa legata a PTEN (PHTS) è 6 anni.
Diagnosi
Non esistono criteri specifici per la diagnosi, che di solito viene posta in base al quadro clinico. Possono essere usati i criteri pediatrici del sistema di punteggio PTEN, basati essenzialmente sulla presenza o assenza della macrocefalia e sulla presenza di uno dei quattro sottocriteri (disturbi dello spettro autistico, segni dermatologici, malformazioni vascolari e/o poliposi gastrointestinale). Una mutazione di PTEN conferma l’appartenenza della BRRS al gruppo delle PHTS.
Diagnosi differenziale
La diagnosi differenziale si pone con:
- sindrome Proteus;
- sindrome da poliposi giovanile;
- sindrome di Lhermitte-Duclos,
- sindrome di Peutz-Jeghers;
- sindrome di Birt-Hogg-Dubé;
- sindrome di Cowden;
- sindrome di Gorlin;
- neurofibromatosi tipo 1.
Diagnosi prenatale
È possibile la diagnosi prenatale nelle gravidanze a rischio, se è nota la mutazione patogenetica in un familiare affetto.
Consulenza genetica
La BRRS ha una trasmissione autosomica dominante. Ai pazienti con mutazioni di PTEN può essere offerta la consulenza genetica e i loro familiari asintomatici dovrebbero essere analizzati per cercare la mutazione e, di conseguenza, identificare coloro che necessitano di essere monitorati prima dell’esordio dei sintomi.
Trattamento
La presa in carico e il trattamento sono multidisciplinari. È molto importante il monitoraggio delle complicazioni della poliposi amartomatosa gastrointestinale, in quanto possono essere più gravi rispetto a quelle osservate nella CS. Una volta identificata una mutazione della linea germinale di PTEN, i pazienti dovrebbero essere sottoposti a screening con l’esame ecografico della tiroide. Nei pazienti prima dei 18 anni si raccomandano la visita dermatologica e l’esame ecografico della tiroide con cadenza annuale. Tra i 35 e i 40 anni, i pazienti possono iniziare a sottoporsi alla colonoscopia e a un imaging renale biennale. Le donne devono effettuare ogni mese l’autopalpazione del seno, una volta l’anno la visita senologica, e dall’età di 30 anni l’ecografia transvaginale o la biopsia dell’endometrio. È inoltre importante prestare attenzione alle malformazioni neurologiche e vascolari, e ai sintomi del grosso intestino.
Prognosi
La prognosi è molto variabile: dipende dal quadro iniziale e dal genotipo.
Leggi anche:
- Polipi intestinali e polipectomia: come si esegue, biopsia e pericoli
- Poliposi gastrointestinale giovanile: cause, sintomi, diagnosi, terapia
- Poliposi infantile e giovanile: cause, sintomi, diagnosi, terapia
- Poliposi adenomatosa familiare: cause, sintomi, diagnosi, terapia
- Sindrome di Peutz-Jeghers: cause, sintomi, diagnosi, terapia
- Polipi intestinali: tipi, cause, fattori di rischio, fattori protettivi
- Polipi intestinali: sintomi, segni, esami, diagnosi, screening
- Polipi intestinali: terapia, chirurgia, colectomia, farmaci, prevenzione
- Si può vivere senza intestino? Colectomia e colostomia
- Stomie: cosa sono, a che servono, quanti tipi esistono?
- Blocco intestinale (occlusione) in bambini e anziani: cause e cure
- Differenze tra ileo meccanico ed ileo paralitico: cause, sintomi e trattamenti
- Quando il sangue esce dall’ano: rettorragia e proctorragia
- Feci nere e melena: cause e cure in adulti e neonati
- Feci con sangue, muco, cibo: quando preoccuparsi?
- Feci dalla bocca: il vomito fecaloide
- Sangue occulto nelle feci: da cosa dipende e come si cura
- Differenza tra polipi e diverticoli: sintomi comuni e diversi
- Differenza tra polipo e tumore
- Colonscopia: cos’è, quando si fa, preparazione e rischi
- Colonscopia tradizionale o colonscopia virtuale?
- Colonscopia: rischi, effetti collaterali e complicanze
- Differenza tra colonscopia e gastroscopia
- Malattia di Crohn: cos’è, cause scatenanti, sintomi, cure e dieta
- Colite ulcerosa: cause, diagnosi, cura, dieta, cosa mangiare, rimedi
- Differenze tra malattia di Crohn e colite ulcerosa: sintomi comuni e diversi
- Visita proctologica: preparazione, costo, procedura, è dolorosa?
- Sangue dall’ano senza defecare: cause, cure quando preoccuparsi?
- Differenza tra ematochezia, rettorragia, proctorragia, melena, sangue occulto nelle feci
- Dolore prima, durante e dopo la defecazione: cause e cure
- Emorroidi interne e esterne: cause, sintomi, cura e rimedi
- Prolasso rettale: intervento, cure, ginnastica, immagini, pomate
- Incontinenza fecale: dieta, Alzheimer, parto, cause e terapie
- Fistola anale e ascesso: sintomi, intervento, cura, immagini
- Cisti pilonidale: cura, crema, intervento, sintomi, rimedi, immagini
- Sindrome dell’intestino irritabile: cause, sintomi e diagnosi
- Cause di pancia gonfia: alimentazione ed emozioni
- Stitichezza acuta e cronica: tipi, cause, trattamenti medici e rimedi
- Feci galleggianti e maleodoranti: cause e quando chiamare il medico
- Colore delle feci: normale e patologico
- Feci pastose e maleodoranti: malassorbimento e cattiva digestione
- Feci gialle, giallo oro, giallastre: cause ed interpretazione clinica
- Coprocoltura feci per salmonella: perché e come si fa
- Carboidrati, proteine e grassi: come vengono assorbiti nell’intestino?
- Differenza tra colite ulcerosa, muco-membranosa, da fermentazione e da putrefazione
- Cosa sono i diverticoli e quali sono i loro sintomi?
- Diverticoli e diverticolosi del colon: cause, sintomi e terapie
- Diverticolo di Meckel: chirurgia, immagini, dieta, terapia
- Tumore dello stomaco: sintomi iniziali, sopravvivenza, cure
- Malattia di Cronkhite-Canada: cause, sintomi, diagnosi, terapia
- Gastrite acuta e cronica: cause, fattori di rischio, sintomi, segni, complicanze
- Gastrite acuta e cronica: esami, diagnosi, terapia, consigli, dieta, cibi da evitare
- Gastrite cronica, quando il bruciore di stomaco non dà tregua
- Acidità di stomaco e bruciore: tutti i farmaci antiacidi
- Bruciore di stomaco: cosa mangiare, come dormire e rimedi naturali
- Acidità di stomaco: come combatterla con i farmaci antiacidi
- Gastrina alta, bassa, valori, funzioni, cos’è e dove è prodotta
- Bruciore retrosternale (pirosi): cause, sintomi, diagnosi, terapie, rischi
- Inibitori della pompa protonica: effetti collaterali e meccanismo d’azione
- I migliori farmaci antiacidi da banco, senza ricetta medica
- Farmaci procinetici: effetti collaterali e meccanismo d’azione
- Si può vivere senza stomaco? Conseguenze della gastrectomia
- Come fare un clistere evacuativo: procedura semplice con enteroclisma
- Fare un clistere evacuativo: procedura semplice con peretta
- Microclisma: cos’è e come si usa in adulti e neonati
- Differenze tra clistere, peretta, enteroclisma, microclisma
- Quando è utile fare un clistere evacuativo?
- Clistere: dopo quanto fa effetto?
- Differenze tra clistere ed enteroclisma
- Fecaloma: tappo di feci durissime, cause, sintomi e rimedi
- Fecaloma ed ostruzione intestinale: quando chiamare il medico
- Feci dure, stitichezza e dolore defecazione: cause e cure
- Come ammorbidire le feci dure in modo naturale e con i farmaci
- Manovre ed altri accorgimenti per facilitare l’evacuazione
- Sindrome da defecazione ostruita: sintomi, cause e terapie
- Di cosa sono fatte le feci?
- Le feci si formano lo stesso quando si è a digiuno?
- Perché le feci hanno un odore cattivo e sgradevole?
- Quanto pesano le feci prodotte in un giorno?
- Dopo quanto tempo il cibo ingerito viene espulso con le feci?
- Le feci più piccole e più grandi del mondo
- Coprofagia umana e animale: cause, rischi e pericoli di ingerire feci
- Si può produrre energia dalle feci?
- Trapianto di feci per clostridium difficile, colite e Morbo di Crohn
- Feci chiare, ipocoliche, acoliche significato e terapie
- Incontinenza feci e gas intestinali: cause, sintomi, rimedi e dieta
- Meteorismo intestinale: cos’è, cause, rimedi, dieta, cibi da evitare
- Defecografia: cos’è, a che serve, come ci si prepara, è dolorosa?
- Come mettere facilmente una supposta a neonati, bambini, adulti
- Reflusso gastroesofageo: sintomi, diagnosi e cura
- Reflusso gastroesofageo: dieta, stress, alcolici, latte e notte
- Reflusso gastroesofageo: terapia farmacologica e chirurgica
- Esofago di Barrett, tumore e reflusso gastroesofageo
- Esofago di Barrett: sintomi iniziali, diagnosi, terapia, dieta e chirurgia
- Differenza tra metaplasia, displasia e neoplasia con esempi
- Sindrome di Zollinger-Ellison (gastrinoma): cause, sintomi, terapia, sopravvivenza
- Gastrite ipertrofica gigante (malattia di Ménétrier): cause, sintomi, diagnosi, cure
- L’apparato digerente: cos’è, com’è fatto, a che serve e come funziona?
- Esofago: anatomia e funzioni in sintesi
- Stomaco: anatomia e funzioni in sintesi
- Capacità massima dello stomaco: si può “mangiare fino a scoppiare”?
- Disfagia nell’anziano: definizione, epidemiologia, cause e fattori di rischio
- Disfagia nell’anziano: sintomi, segni, diagnosi, esami, trattamento
- Differenza tra disfagia di tipo ostruttivo e di tipo motorio
- Differenza tra disfagia ai liquidi e ai solidi
- Differenza tra disfagia ed odinofagia: cause comuni e diverse
- Differenza tra disfagia orofaringea ed esofagea: sintomi comuni e diversi
- Differenza tra disfagia ostruttiva ed occlusione intestinale
- Alimentazione e disfagia nel paziente con morbo di Parkinson
- Polmonite ab ingestis: cause, tempi di guarigione, morte, sopravvivenza
- Inalazione di cibo e corpi estranei nelle vie aeree: sintomi e cosa fare
- Morte per soffocamento: segni, sintomi, fasi e tempi
- Soffocamento da cibo, liquidi, saliva in bimbi e adulti: cosa fare?
- Acalasia esofagea: cause, sintomi, cure e prevenzione
- Ritirato in tutta Italia il famoso farmaco anti reflusso: ecco i lotti interessati
- Manometria esofagea: preparazione, esecuzione, rischi, prezzo
- Manometria esofagea 24H: cos’è, a che serve, quando è necessaria
- Manometria esofagea ad alta risoluzione (HRM): cos’è, a che serve, quando è necessaria
- pH-metria esofagea e gastrica: preparazione, esecuzione, rischi, prezzo
- Laringoscopia diretta e indiretta: anestesia, costo, è dolorosa?
- L’esofagogastroduodenoscopia: cos’è, preparazione, è dolorosa o pericolosa?
- Colangiopancreatografia retrograda (ERCP): cos’è, preparazione, è dolorosa o pericolosa?
- Differenza tra ulcera gastrica, duodenale, peptica ed esofagea
- Ulcera peptica: complicanze, cura, dieta, quando è pericolosa
- Cosa succede al cibo nello stomaco dopo averlo ingerito?
- Peristalsi intestinale ed antiperistalsi: caratteristiche e funzioni
- Fundoplicatio secondo Nissen-Rossetti: intervento e rischi
- Acido cloridrico e succo gastrico dello stomaco: di cosa è fatto ed a che serve
- Meccanismi e controllo della secrezione acida dello stomaco
- Dispepsia: cos’è, sintomi, come si fa la diagnosi e terapia
- Stomaco: come fa a digerire il cibo che mangi ed a dirti che sei “pieno”
- Come vincere l’ansia per evitare di mangiare fuori pasto
- Eliminare la tensione nervosa allo stomaco con i rimedi naturali
- Incontinenza fecale primitiva e secondaria: cos’è e come si cura
- Vomitare sangue ed ematemesi: cos’è, cosa fare, cause e terapie
- Da cosa viene causata l’ulcera allo stomaco?
- Breath test Helicobacter: come funziona, come si fa e valori
- Infezione da Helicobacter Pylori: cosa causa, come si riconosce e cura
- Classifica dei cibi con maggior quantità di fibre esistenti
- Fibre: definizione, fabbisogno, integratore, alimenti, carenza
- Antiossidanti: alimenti ed integratori migliori contro i radicali liberi
- Sali minerali: definizione, funzioni, alimenti, integratori [GUIDA COMPLETA]
- Differenza tra tumore benigno, maligno, neoplasia, cancro e metastasi
- Cos’è un tumore? Perché viene il cancro? Quali sono le cause?
- Come nasce un cancro? Cosa sono i cancerogeni e come avviene la cancerogenesi?
- Come prevenire i tumori ed il cancro? I 10 cambiamenti consigliati
- Differenza tra tumore e tessuto normale con esempi
- Cosa sono le metastasi? Tutti i tumori danno metastasi?
- Che significa malattia terminale?
- Stadiazione e classificazione TNM: cancro curabile o terminale?
- Cancro dell’esofago: epidemiologia, età di esordio, tipologie
- Cancro dell’esofago: cause, fattori di rischio, esofago di Barrett
- Cancro dell’esofago: sintomi e segni iniziali, tardivi e di metastasi
- Cancro dell’esofago: esami, diagnosi, stadiazione, classificazione TNM, gravità
- Cancro dell’esofago: trattamento, chirurgia, chemioterapia, radioterapia
- Cancro dell’esofago: prognosi, mortalità, sopravvivenza, aspettativa di vita
- Cancro dell’esofago: prevenzione, dieta, cibi da evitare, consigli
- Cancro dello stomaco: epidemiologia, età di esordio, tipologie
- Cancro dello stomaco: cause, fattori di rischio, fattori protettivi
- Cancro dello stomaco: sintomi e segni iniziali, tardivi e di metastasi
- Cancro dello stomaco: esami, diagnosi, stadiazione, classificazione TNM, gravità
- Cancro dello stomaco: trattamento, chirurgia, chemioterapia, radioterapia
- Cancro dello stomaco: prognosi, mortalità, sopravvivenza, aspettativa di vita
- Cancro dello stomaco: prevenzione, dieta, cibi da evitare, consigli
- Carcinoma precoce dello stomaco (early gastric cancer): cause, sintomi, diagnosi, cure
- Tumore del colon retto: sintomi iniziali, tardivi e ritardo nella diagnosi
- Tumore del colon retto: diagnosi, metastasi, prognosi e stadiazione
- Tumore del colon retto: trattamento chirurgico, radioterapia e chemioterapia
- Tumore del colon retto con metastasi: chirurgia, chemioterapia e terapie biologiche
- Tumore del colon retto: terapia personalizzata col test RAS
- Esofagite: cause, cronica, sintomi, cura, dieta, alimentazione
- Esofagite da reflusso: cause e fattori di rischio
- Esofagite da reflusso: sintomi e segni
- Esofagite da reflusso: esami, diagnosi, endoscopia, breath test
- Esofagite da reflusso: gravità e complicanze
- Esofagite da reflusso: terapia, farmaci, chirurgia
- Esofagite da reflusso: consigli, dieta, cibi da evitare, come dormire, prevenzione
- Reflusso gastro-esofageo nell’anziano: epidemiologia, cause, fattori di rischio
- Reflusso gastro-esofageo nell’anziano: sintomi e segni
- Reflusso gastro-esofageo nell’anziano: esami, diagnosi, gravità
- Reflusso gastro-esofageo nell’anziano: complicanze, prevenzione, dieta
- Reflusso gastro-esofageo nell’anziano: terapia a breve termine, farmaci, interazioni
- Reflusso gastro-esofageo nell’anziano: terapia a lungo termine, chirurgia, farmaci
Lo Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o unisciti al nostro gruppo Facebook o ancora seguici su Twitter, su Instagram o su Pinterest, grazie!
