
 Con “acondrogenesi” (in inglese “achondrogenesis”) si indica un gruppo di patologie che corrispondono alle forme più gravi di “condrodisplasia congenita” (caratterizzata da malformazione delle ossa e della cartilagine). Queste condizioni sono caratterizzate da un piccolo corpo, arti corti e altre anomalie scheletriche. L’acondrogenesi è una malattia rara, letale già in epoca prenatale (prima ancora del parto), con aspettativa di vita praticamente nulla e caratterizzata da un difetto nell’ossificazione endocondrale.
Con “acondrogenesi” (in inglese “achondrogenesis”) si indica un gruppo di patologie che corrispondono alle forme più gravi di “condrodisplasia congenita” (caratterizzata da malformazione delle ossa e della cartilagine). Queste condizioni sono caratterizzate da un piccolo corpo, arti corti e altre anomalie scheletriche. L’acondrogenesi è una malattia rara, letale già in epoca prenatale (prima ancora del parto), con aspettativa di vita praticamente nulla e caratterizzata da un difetto nell’ossificazione endocondrale.
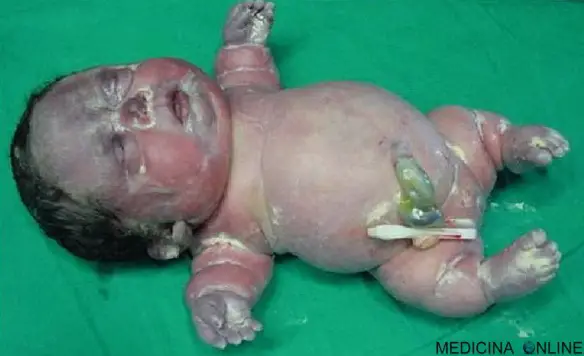
Nella foto in alto potete vedere una bimba con acondrogenesi di tipo I subito dopo la nascita: pesava 1810 grammi, misurava 31 centimetri e purtroppo è deceduta circa 30 minuti dopo essere venuta al mondo.
Tipologie
Si conoscono due tipi principali di acondrogenesi, la cui differenziazione è difficile in base ai soli dati clinici e radiologici. Esistono poi il tipo 1A e il tipo 1B. Questi tipi si distinguono per i loro segni e sintomi, pattern di ereditarietà e causa genetica. Potrebbero esistere altri tipi di acondrogenesi, ma non sono stati ancora ben studiati.
Sintomi e segni
Le caratteristiche cliniche comprendono il nanismo ad arti corti con tronco corto, addome prominente, con cranio sproporzionatamente grande e anasarca. Il quadro radiologico è caratteristico, con virtuale assenza di ossificazione della colonna vertebrale, dell’osso sacro e delle ossa del bacino.
Trasmissione
La malattia è causata da mutazioni geniche trasmesse in modo autosomico dominante o recessivo in base al tipo. L’acondrogenesi tipo 1A è trasmessa come mutazione autosomica recessiva: i genitori sono portatori sani del gene mutato e hanno il 25% di probabilità di trasmettere la malattia a ciascuno dei figli.
Il sottotipo 1B è causato da mutazioni nel gene della displasia diastrofica, SLC26A2 (DTDS), un trasportatore dei solfati; l’acondrogenesi tipo 2 è causata da mutazioni dominanti de novo nel gene del collagene tipo II-1, COL2A1.
Eziopatologia
L’acondrogenesi di tipo 1A è causata da un difetto nei microtubuli dell’apparato di Golgi. Nei topi, una mutazione non senso nel gene dell’interferone 11 recettore dell’ormone tiroideo (Trip11), che codifica per la proteina 210 associata al microtubulo di Golgi (GMAP-210), ha provocato difetti simili alla malattia umana. Quando il loro DNA è stato sequenziato, i pazienti umani con acondrogenesi di tipo 1A avevano anche mutazioni con perdita di funzione in GMAP-210. GMAP-210 sposta le proteine dal reticolo endoplasmatico all’apparato di Golgi. A causa del difetto, GMAP-210 non è in grado di spostare le proteine e rimangono nel reticolo endoplasmatico, che si gonfia. La perdita della funzione dell’apparato di Golgi colpisce alcune cellule, come quelle responsabili della formazione di ossa e cartilagini, più di altre.
Diagnosi
La diagnosi viene effettuata alla nascita sulla base delle caratteristiche cliniche, che sono subito ben evidenti, e sull’osservazione radiografica.
Diagnosi prenatale
Durante la gravidanza il sospetto di acondroplasia si avanza in presenza di particolari anomalie del feto evidenti all’ecografia circa dalla 14ª settimana (femori molto corti, ridotta ossificazione dei corpi vertebrali e delle ossa degli arti). In alcuni casi è possibile effettuare la diagnosi prenatale con indagine sui villi coriali; questo è possibile in genere nelle gravidanze successive dopo un primo caso di malattia.
Terapie
Attualmente non esiste alcuna terapia per l’acondrogenesi.
Prognosi
A causa dei loro seri problemi di salute, i neonati con acondrogenesi di solito nascono prematuri e muoiono poco dopo la nascita per insufficienza respiratoria, oppure nascono già morti.
Per approfondire:
- Nanismo: sintomi, cura, cause, terapia, diagnosi e prevenzione
- Acondroplasia: diagnosi prenatale ecografica e aspettativa di vita
- Altezza: quando si può parlare di nanismo o gigantismo
- L’altezza media italiana nel 2017 di maschi e femmine
- Charlotte, la bambina più piccola del mondo
Leggi anche:
- Osteogenesi imperfetta (malattia delle ossa di cristallo): cause, sintomi e cure
- Ittiosi: cos’è, sintomi, cure, come si trasmette, è contagiosa?
- Ittiosi Arlecchino: diagnosi prenatale, aspettativa di vita, immagini, video
- Ipotelorismo interpupillare: quando gli occhi sono troppo vicini tra loro
- Ipertelorismo interpupillare e mammellare: sintomi, cause, cure
- Telecanto: cause, sintomi, terapia, etimologia
- Ereditarietà X linked, malattie dominanti e recessive legate al cromosoma X: significato, esempi
- Differenza tra autosomica dominante e recessiva con esempi
- Aplasia cutanea circoscritta del vertice: cause, sintomi, cure
- Sindrome di Noonan: cause, sintomi nel neonato, aspettative di vita
- Sindrome di Down: cause, sintomi in gravidanza e nei neonati
- Sindrome di Turner: cariotipo, cause, sintomi e segni caratteristici
- Sindrome di Klinefelter: cariotipo, cause, sintomi e cura
- Sindrome di Möbius: cause, sintomi, diagnosi e terapia
- Sindrome di Pierre Robin: cause, sintomi, diagnosi, cure, prognosi
- Sindrome di Stickler: cause, sintomi, diagnosi, cure, prognosi
- Sindrome di Pfeiffer: cause, sintomi, diagnosi, cure, prognosi
- Fibrosi cistica polmonare: cos’è, sintomi in neonati e bambini, cure
- Malattia di Huntington: cos’è, ereditarietà, come si trasmette, età di insorgenza
- Anemia falciforme: cosa significa, cause, sintomi e cure
- Differenze tra la distrofia muscolare di Duchenne e di Becker
- Differenza tra distrofia muscolare e sclerosi multipla
- Differenza tra distrofia muscolare ed atrofia muscolare progressiva
- Differenza tra distrofia muscolare e SLA (sclerosi laterale amiotrofica)
- Talassemia: cos’è, sintomi, cure, differenti tipi ed alimentazione
- Anemia da carenza di ferro: cause, sintomi e cure
- Emocromo completo con formula leucocitaria: valori, interpretazione e significato
- Ittero emolitico, colestatico, ostruttivo, neonatale: significato, occhi, cura
- Aracnodattilia, segno del pollice e del polso, Sindrome di Marfan e di Beals
- Ectrodattilia: cause, cure ed immagini
- Polidattilia; cause, ereditarietà, sindromica e chirurgia
- Sindrome di Beals e padiglione auricolare “accartocciato”: sintomi e cure
- Diabete insipido: cause, diagnosi e trattamento
- Malattia granulomatosa cronica: causa, trasmissione, sintomi, cura
- Distrofia muscolare in adulti e bambini: sintomi, cause, diagnosi e cure
- Differenze tra la distrofia muscolare di Duchenne e di Becker
- Emofilia: cos’è, diagnosi, sintomi, tipi, terapia e cura
- Deficit di glucosio-6-fosfato deidrogenasi e favismo: cause, sintomi, terapie
- Sindrome di Wiskott-Aldrich: cause, sintomi, trasmissione, cure
- Sindrome di Lesch-Nyhan: cause, sintomi, trasmissione, diagnosi, cure
- Sindrome dell’X fragile in uomini e donne: sintomi, aspettativa di vita, cure
- Autismo: definizione, cause, sintomi, diagnosi e cure
- Sindrome di Asperger in bambini ed adulti: primi sintomi, terapie
- Sindrome di Rett: cause, sintomi, tipi, diagnosi, stadi, cure, morte
- Disturbo disintegrativo dell’infanzia (demenza infantile): cause, sintomi e terapie
- Differenza tra polidattilia, sindattilia, ectrodattilia, oligodattilia e aracnodattilia
- Cos’è un cromosoma ed a che serve?
- Sindrome dell’idiota sapiente: cause, caratteristiche e sintomi
- Sindrome del tramonto o del crepuscolo: cause, sintomi e cura
- Ritardo mentale nei bambini lieve, moderato, grave: si guarisce?
- Che cos’è l’intelligenza umana: definizione, significato e psicologia
- Quoziente d’intelligenza: valori, significato, test ed ereditarietà
- Problem solving: cos’è, caratteristiche, tecniche, fasi ed esempi
- Sindrome di Tourette: cause, sintomi, diagnosi e trattamento
- Sindrome di Tourette: si può guarire definitivamente? Come si guarisce?
- Disturbi pervasivi dello sviluppo (disturbi dello spettro autistico)
- Disturbi specifici di apprendimento (DSA): definizione, cause, sintomi, cure
- Dislessia: cos’è, come riconoscerla, come affrontarla e superarla
- Il disagio psicologico nel bambino con dislessia
- Epilessia: riconoscere in tempo l’arrivo di una crisi e come comportarsi
- Epilessia infantile: come comportarsi col proprio figlio?
- Si può morire di epilessia?
- Disgrafia: esempi, come riconoscerla precocemente, test, rimedi
- Disortografia: cause, come riconoscerla, esempi, rimedi, si guarisce?
- Differenze tra disgrafia e disortografia
- Discalculia: significato, tipi, sintomi, diagnosi e terapia
- Disprassia a scuola: sintomi, esercizi, si guarisce?
- Disturbo specifico della compitazione: significato, sintomi, cure
- Disturbo specifico del linguaggio: sintomi, classificazione, si guarisce?
- Ritardo semplice di linguaggio: definizione e classificazione
- Balbuzie e disfluenze: significato, cause, sintomi, rimedi
- Differenza tra allele dominante e recessivo
- Differenza tra omozigote ed eterozigote
- Differenza tra gene e allele
- Differenza tra genotipo e fenotipo
Lo Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o unisciti al nostro gruppo Facebook o ancora seguici su Twitter, su Instagram o su Pinterest, grazie!
