
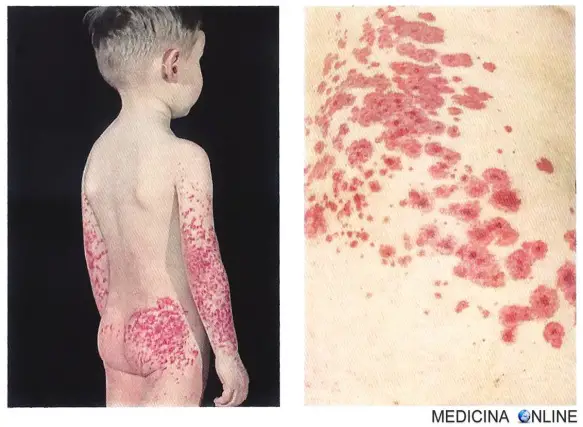
Porpora di Shöenlein-Henoch, tipica localizzazione intrafocale delle petecchie
Alcune vasculiti ad eziologia ancora non del tutto chiara, probabilmente riconoscono una patogenesi immunitaria. Tale ipotesi si basa sulle analogie dei reperti immunoistochimici con quelli osservati in animali sensibilizzati e sulla frequente associazione di queste sindromi vascolari con manifestazioni allergiche ben definite, sostenute da allergeni diversi, ma soprattutto da farmaci (penicillina
ed altri antibiotici, sulfamidici, sieri eterologhi, etc.).
Sulla base dei diversi quadri clinici (vario interessamento di organi e di vasi, esistenza o meno di danno renale, di ipertensione arteriosa e di eosinofilia) ed istologici (necrosi e degenerazione fibrinoide, eventuale presenza di granulo mi extravascolari), sono state descritte forme morbose distinte, la cui patogenesi ed i cui rapporti reciproci non sempre risultano ben delineati. Rientrano nel capitolo delle vasculiti da ipersensibilità, i cui limiti sono ancora molto vaghi, la malattia da siero, la porpora di Shöenlein-Henoch (o “porpora reumatoide”), la vasculite allergica, la vasculite orticarioide, l’angioite necrotizzante da ipersensibilità, la granulomatosi di Wegener e l’arterite giganto cellulare.
Caratteristiche comuni
Le caratteristiche comuni delle vasculiti da ipersensibilità sono rappresentate dal coinvolgimento dei vasi di piccolo calibro e dal prevalente interessamento cutaneo, con quadri di porpora e/o di orticaria. Sono anche presenti, frequentemente, sintomi generali (febbre, mialgie, malessere generale, etc.) o segni di interessamento specifico di organi
(adenopatie, epatosplenomegalia, manifestazioni respiratorie, gastrointestinali, neurologiche, etc.).
Criteri diagnostici
Per l’inquadramento di una vasculite nel gruppo nosologico delle vasculiti da ipersensibilità debbono essere soddisfatti almeno tre dei seguenti criteri:
- comparsa della sintomatologia in età superiore ai 16 anni;
- dato anamnestico dell’assunzione di farmaci in grado di innescare l’esordio della sindrome clinica;
- presenza di elementi purpurici palpabili in assenza di piastrinopenia;
- presenza di manifestazioni esantematiche maculopapulari;
- reperti bioptici di granulociti neutrofili ed eosinofili a localizzazione intravasale (pareti venulari e/o arteriolari) e perivasale.
Patogenesi
Il danno vascolare ed endoteliale è dovuto essenzialmente alla deposizione di immunocomplessi nelle pareti vasali, con conseguente attivazione del sistema complementare. Un notevole ruolo patogenetico è svolto dalle molecole di adesione (ICAM-1, ELAM-1, VCAM-1, etc.), che favoriscono l’adesione e l’attivazione dei leucociti.
Deve essere ricordato che anticorpi anti-endotelio (AECA), anticorpi anti-citoplasma dei neutrofili (ANCA), anticorpi linfocitotossici (LTA) ed anticorpi anti-membrane basali sono rilevabili nel siero di pazienti affetti da forme diverse di vasculite; alcuni di questi anticorpi, in particolare gli AECA, sono in grado di mediare, in presenza di mononucleati, una citotossicità anticorpo-dipendente.
Anticorpi anti-citoplasma dei neutrofili (ANCA)
Per quanto riguarda gli ANCA, se ne distinguono, a seconda dei pattern rilevati all’immunofluorescenza indiretta, due tipi principali:
- c-ANCA, di tipo “citoplasmatico” o “classico”, rivolti verso la proteinasi 3 (solo eccezionalmente verso la mieloperossidasi) ed altamente specifici per la granulomatosi di Wegener (80-95% di prevalenza in fase attiva di malattia).
- p-ANCA, di tipo “perinucleare”, diretti contro la mieloperossidasi e più di rado contro altri enzimi granulocitari (elastasi, lactoferrina, catepsina G, lisozima, etc.), che risultano correlati ad altre vasculiti.
Anticorpi anti-endotelio (AECA)
Gli AECA, che sono di classe IgG o IgM (raramente IgA), possono riconoscere antigeni “costitutivi” (espressi sulle cellule endoteliali quiescenti) o “inducibili” (espressi sulle cellule endoteliali preattivate con citochine). Va rilevato che gli AECA possono ritrovarsi anche in altre malattie, a patogenesi immunologica (LES, artrite reumatoide, etc.) o non immunologica (infarto miocardico, etc.), e, sia pure a basso titolo, in individui apparente-
mente sani, in assenza di processi vasculitici.
Porpora di Shöenlein-Henoch
In alcuni casi di porpora di Schònlein-Henoch, che colpisce prevalentemente soggetti in età pediatrica, è stato sospettato il ruolo di vari farmaci (ampicillina, penicillina, eritromicina, chinolonici, clorpromazina, etc.). Le manifestazioni cliniche di questa sindrome, caratterizzata dalla presenza di immunocomplessi IgA1 e IgA2 in circolo e nella parete dei vasi dei tessuti interessati, sono rappresentate da lesioni purpuriche (localizzate agli arti inferiori, ma anche a quelli superiori, alle regioni glutee e all’addome, vedi immagine in alto nell’articolo), edema degli arti inferiori, artralgie (in oltre il 70% dei casi). Relativamente frequente è l’interessamento renale (una glomerulonefrite si riscontra nel 20-30% dei casi) e gastrointestinale (emorragie intesti-
nali nel 5-10% dei casi). Istologicamente si dimostra la presenza di una
vasculite necrotizzante. La prognosi è complessivamente buona; tuttavia, in una piccola percentuale di casi (1-2) residua un danno renale permanente.
Sindrome di Churg-Strauss
Una forma particolare di vasculite necrotizzante disseminata si può riscontrare in alcuni casi di asma bronchiale, caratterizzati da intensa eosinofilia ed elevati livelli di IgE totali (sindrome di Churg-Strauss). L’esame istologico dimostra la presenza di granulo mi eosinofili ed epitelioidi, con necrosi fibrinoide.
È presente anche un’infiltrazione eosinofila a carico di vari organi ed apparati, soprattutto a livello broncopolmonare (con manifestazioni cliniche asmatiche ovvero del tipo sindrome di Loffler o del tipo polmonite cronica eosinofila) e gastroenterico (gastroenterite eosinofila, con episodi ricorrenti di dolori addominali e di diarrea). Una sindrome broncoasmatica precede sempre, anche di molti anni, le manifestazioni vasculitiche. Le crisi asmatiche, che nelle fasi iniziali della malattia sono in genere lievi, divengono progressivamente più frequenti ed intense e risentono scarsamente della terapia con broncodilatatori e con glicocorticoidi per via inalatoria, per cui necessitano di un trattamento con glicocorticoidi per via generale. Dopo questo periodo prodromico, che può durare persino decenni, si assiste alla comparsa di una vasculite sistemica necrotizzante, che interessa soprattutto le piccole arterie (coronariche, cutanee, renali, etc.). Nel sangue periferico si reperta, come riferito in precedenza, una marcata
eosinofilia, che può raggiungere valori estremamente elevati; nel siero, inoltre, si dimostrano elevate concentrazioni di proteina cationica eosinofilo-derivata (ECP). Alti livelli di eosinofili si rinvengono anche nel BAL e nelle secrezioni nasali. Oltre ad anticorpi anti-nucleari, si evidenziano spesso anticorpi anti-citoplasma dei neutro fili (ANCA), che possono costituire marker di attività.
La sindrome di Churg-Strauss è molto sensibile al trattamento con dosi elevate di glicocorticoidi, che però determinano una remissione soltanto temporanea, con netta diminuzione dell’eosinofilia e dei livelli di ECP sierici; successivamente, un graduale incremento dell’ eosinofilia precede quasi sempre la ricomparsa delle manifestazioni cliniche, con prognosi sfavorevole quando si instauri una vasculite sistemica.
Per approfondire:
- Vasculiti di grandi, medi e piccoli vasi: tipi, cause, sintomi, cure
- Arteriti: significato, cause, sintomi, diagnosi, rischi, terapie
Leggi anche:
- Visita allergologica: svolgimento, esami, preparazione, durata, costo
- Eruzioni esantematiche, eritematose, vescicolo-bollose e pigmentarie da farmaci
- Differenza tra allergia, pseudoallergia e intolleranza alimentare
- Consigli per la prevenzione delle allergie inalatorie da pollini
- Consigli per la prevenzione delle allergie da alimenti e additivi alimentari
- Consigli per la prevenzione delle allergie da acari e miceti (funghi)
- Consigli per la prevenzione delle dermatiti da contatto (lattice, nichel, cosmetici…)
- Consigli per la prevenzione delle allergie da derivati animali (cani, gatti, volatili…)
- Consigli per la prevenzione di allergie da imenotteri (api e vespe)
- Consigli per la prevenzione di allergie da farmaci e pseudoallergie
- Orticaria acuta e cronica: cause, sintomi, diagnosi, immagini, cura, rimedi
- Angioedema: cause, sintomi, immagini, cura, consigli, rimedi naturali
- Malattia da siero: cause, sintomi, complicanze, diagnosi e terapie
- Congiuntivite allergica primaverile e altre patologie oculari da allergia
- Sindromi neuroallergiche: emicrania da allergia, cefalea istaminica e da glutammato sodico
- Differenza tra orticaria e angioedema
- Sindrome orticaria-angioedema: cause, sintomi, diagnosi, immagini, cura, rimedi
- Dermatiti esfoliative da farmaci: la sindrome di Lyell
- Segno Asboe-Hansen: indicazioni, esecuzione, positivo
- Shock anafilattico (shock allergico): cause, fattori di rischio, patogenesi
- Shock anafilattico: sintomi, diagnosi, prognosi, gravità, morte
- Shock anafilattico: terapia, cosa fare, consigli per prevenirlo
- Esami allergologici di primo, secondo e terzo livello per la diagnosi di allergia
- Prick test in bambini e adulti: esecuzione, allergeni, controindicazioni, rischi
- Patch test: preparazione, esecuzione, rischi, allergeni, costo
- Prist test in adulti e bambini, valori normali e patologici, interpretazione
- Rast test: preparazione, esecuzione, rischi, allergeni, costo
- Test di provocazione in medicina: cosa sono, a che servono, come si svolgono?
- Test di provocazione bronchiale con metacolina: esecuzione, preparazione, rischi
- Iper-reattività bronchiale: significato, sintomi, diagnosi e cure
- Test di eliminazione e diete oligoallergeniche in allergologia
- Reazioni indesiderate durante i test allergologici: frequenza, tipi, cosa fare, prevenzione
- Dermatite da contatto: cause, fattori predisponenti, allergeni, patogenesi
- Dermatite da contatto: sintomi e segni di fase acuta, subacuta e cronica
- Dermatite da contatto: diagnosi, esami, terapie e consigli
- Allergia al latte vaccino in neonati e bambini
- Asma bronchiale estrinseca, intrinseca, occupazionale, stabile: cause, sintomi, cure
- Consigli per la prevenzione delle sindromi asmatiche
- Attività fisica e asma bronchiale: sport consigliati e sconsigliati
- Allergeni ed epitopi: definizione, classificazione, elenco, tipi
- Differenza tra le reazioni di ipersensibilità di tipo I, II, III, IV e V
- Immunoterapia specifica (iposensibilizzazione): indicazioni, controindicazioni
- Immunoterapia specifica (iposensibilizzazione): vie di somministrazione dell’allergene
- Immunoterapia specifica (iposensibilizzazione): estratti allergenici, miscele, conservazione
- Immunoterapia specifica: metodiche di somministrazione, dosaggi
- Immunoterapia specifica: epoca d’inizio, schemi di trattamento, durata, sospensione
- Immunoterapia specifica: schemi non convenzionali, iposensibilizzazione a farmaci e veleni di imenotteri
- Immunoterapia specifica: monitoraggio e valutazione dell’efficacia della terapia
- Immunoterapia specifica: reazioni indesiderate locali, sistemiche, gravi
- Test per allergie e intolleranze alimentari
- Differenza tra reazione allergica, anafilassi e shock anafilattico
- Differenze tra allergia alimentare ed intolleranza alimentare
- Dermatite atopica: cause, sintomi e terapie di una patologia della pelle molto diffusa
- Dermatite atopica: giusta alimentazione e probiotici quando le cure tradizionali falliscono
- Dermatite seborroica: cause, immagini, cure, rimedi naturali, shampoo consigliato
- Pelle arrossata ed irritata a causa del sudore: come curare l’irritazione cutanea?
- Irritazione cutanea in neonati, bambini e gravidanza: i soggetti più a rischio
- Pelle arrossata ed irritata: cause patologiche e non patologiche
- Pelle arrossata ed irritata a causa del sudore: i rimedi naturali
- Pelle arrossata ed irritata: ecco quali farmaci usare
- Morire per una puntura di vespa: capire cos’è uno shock anafilattico può salvarti la vita
- Puntura di vespa: dopo quanto tempo si verifica lo shock anafilattico?
- Puntura di vespa e shock anafilattico: cosa fare e cosa NON fare?
- Puntura di vespa e shock anafilattico: cosa fare prima dell’arrivo dell’ambulanza?
- Cos’è l’adrenalina ed a cosa serve?
- Asma bronchiale estrinseca, intrinseca, occupazionale, stabile: cause, sintomi, cure
- Asma bronchiale in bambini e adulti: cause, sintomi e cura
- Asma bronchiale: spirometria e diagnosi differenziale
- Asma bronchiale: fattori di rischio ambientali e genetici
- Asma occupazionale: cause, sintomi, diagnosi e terapie
- Malattie respiratorie occupazionali o professionali: effetti, elenco, tipi
- Spirometria diretta ed indiretta: come si esegue ed a cosa serve
- Deficit di glucosio-6-fosfato deidrogenasi e favismo: cause, sintomi, terapie
- L’incredibile allergia di Ashleigh Morris
- La grave allergia alle arachidi di Oli Weatherall: perfino un bacio può ucciderlo
- Famosa marca di pennarelli provoca allergie: non usateli!
- Differenza tra latte, lattosio e rispettive intolleranze
- Celiachia, cos’è il glutine, in quali alimenti è contenuto ed in quali no?
- Che significa malattia autoimmune? Spiegazione ed esempi
- Intolleranza al lattosio in adulti e bambini: sintomi e test
- Lattosio: cos’è, in quali alimenti si trova ed in quali no
- Lattuga ed altre verdure: contengono lattosio o no?
- Test del respiro (breath test) per intolleranze alimentari: cos’è e come si svolge
- Reazioni avverse ai farmaci: effetti collaterali, tossicità, allergie, idiosincrasie
- Intolleranza al sorbitolo: sintomi, diagnosi, dove si trova il sorbitolo?
- Superare le intolleranze alimentari con la dieta di eliminazione? Meglio lo schema di rotazione dei cibi
- Vaccini: servono davvero? Tutte le verità scientifiche
- Acne cistica: cura definitiva, alimentazione, testosterone
Lo Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o unisciti al nostro gruppo Facebook o ancora seguici su Twitter, su Instagram o su Pinterest, grazie!
