
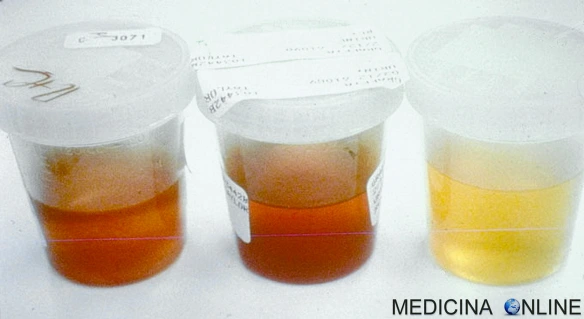
 L’alcaptonuria (anche chiamata “ocronosi endogena” od “ocronosi congenita” o “malattia delle urine scure“; in inglese “alkaptonuria“) è una rara malattia genetica ereditaria recessiva causata da una mutazione nel gene HGD per l’enzima omogentisato 1,2-diossigenasi e che coinvolge il metabolismo della fenilalanina e della tirosina. Il difetto nella funzione dell’enzima omogentisato 1,2-diossigenasi determina un accumulo nel sangue e nelle urine dell’acido omogentisico (e della sua forma ossidata, detta “alcaptone“). L’acido omogentisico e la sua forma ossidata alcaptone vengono escreti nelle urine, conferendogli un colore insolitamente scuro. L’accumulo di acido omogentisico provoca inoltre danni alle cartilagini (che portano all’artrosi) e alle valvole cardiache, oltre a precipitare come calcoli renali e calcoli in altri organi. La malattia è congenita, cioè già presente alla nascita, ma i sintomi di solito si sviluppano nelle persone di età superiore ai 30 anni, sebbene la colorazione scura delle urine sia presente sin dalla nascita. Oltre al trattamento delle complicanze (come il sollievo dal dolore e la sostituzione dell’articolazione per il danno alla cartilagine), è stato scoperto che il farmaco nitisinone sopprime la produzione di acido omogentisico.
L’alcaptonuria (anche chiamata “ocronosi endogena” od “ocronosi congenita” o “malattia delle urine scure“; in inglese “alkaptonuria“) è una rara malattia genetica ereditaria recessiva causata da una mutazione nel gene HGD per l’enzima omogentisato 1,2-diossigenasi e che coinvolge il metabolismo della fenilalanina e della tirosina. Il difetto nella funzione dell’enzima omogentisato 1,2-diossigenasi determina un accumulo nel sangue e nelle urine dell’acido omogentisico (e della sua forma ossidata, detta “alcaptone“). L’acido omogentisico e la sua forma ossidata alcaptone vengono escreti nelle urine, conferendogli un colore insolitamente scuro. L’accumulo di acido omogentisico provoca inoltre danni alle cartilagini (che portano all’artrosi) e alle valvole cardiache, oltre a precipitare come calcoli renali e calcoli in altri organi. La malattia è congenita, cioè già presente alla nascita, ma i sintomi di solito si sviluppano nelle persone di età superiore ai 30 anni, sebbene la colorazione scura delle urine sia presente sin dalla nascita. Oltre al trattamento delle complicanze (come il sollievo dal dolore e la sostituzione dell’articolazione per il danno alla cartilagine), è stato scoperto che il farmaco nitisinone sopprime la produzione di acido omogentisico.
Cenni storici
L’alcaptonuria era una delle quattro malattie descritte da Archibald Edward Garrod, come il risultato dell’accumulo di intermedi dovuto a carenze metaboliche. Garrod ha collegato l’ocronosi con l’accumulo di alcaptani nel 1902; le sue opinioni sull’argomento, inclusa la sua modalità di eredità, sono state riassunte in una Croonian Lecture del 1908 presso il Royal College of Physicians. L’alcaptonuria fu studiata anche da William Bateson nel 1902. Il difetto è stato ristretto al deficit di acido omogentisico ossidasi in uno studio pubblicato nel 1958. La base genetica dell’alcaptonuria è stata chiarita nel 1996, quando sono state dimostrate le mutazioni HGD. Uno studio del 1977 ha dimostrato che una mummia egiziana ocronotica aveva probabilmente sofferto di alcaptonuria.
Diffusione
L’alcaptonuria è una malattia rara; nella maggior parte dei gruppi etnici, la prevalenza dell’alcaptonuria è compresa tra 1:100.000 e 1:250.000. In Slovacchia e nella Repubblica Dominicana la malattia è molto più comune, con una prevalenza stimata in 1:19.000 persone. Per quanto riguarda la Slovacchia, questo non è il risultato di una singola mutazione, ma di un gruppo di 12 mutazioni in specifici punti del gene HGD. Il raggruppamento slovacco è sorto probabilmente in una piccola area nel nord-ovest del paese e si è diffuso dopo gli anni ’50 a causa della migrazione.
Cause e fisiopatologia
L’alcaptonuria ha cause genetiche. Tutte le persone portano nel proprio DNA due copie (una ricevuta da ciascun genitore) del gene HGD, che contiene l’informazione genetica per produrre l’enzima omogentisato 1,2-diossigenasi (HGD) che normalmente si trova in numerosi tessuti dell’organismo ( fegato, reni, intestino tenue, colon e prostata). Nelle persone con alcaptonuria, essendo una malattia genetica recessiva, entrambe le copie del gene contengono anomalie che indicano che il corpo non è in grado di produrre un enzima adeguatamente funzionante. Le mutazioni HGD si trovano generalmente in alcune parti (esoni 6, 8, 10 e 13), ma sono state descritte un totale di oltre 100 anomalie in tutto il gene. Il normale enzima HGD è un esamero (ha sei subunità) che sono organizzati in due gruppi di tre (due trimeri) e contiene un atomo di ferro. Diverse mutazioni possono influenzare la struttura, la funzione o la solubilità dell’enzima. Molto raramente la malattia sembra essere trasmessa con modalità autosomica dominante, in cui una singola copia anormale di HGD da un solo genitore è associata ad alcaptonuria; altri meccanismi o difetti in altri geni potrebbero essere responsabili in questi casi. L’enzima HGD è coinvolto nel metabolismo degli amminoacidi aromatici fenilalanina e tirosina. Normalmente, questi entrano nel flusso sanguigno attraverso il cibo contenente proteine e il ricambio naturale delle proteine nel corpo. La tirosina è specificatamente richiesta per una serie di funzioni, come gli ormoni (ad esempio la tiroxina, l’ormone tiroideo), la melanina (il pigmento scuro della pelle e dei capelli) ed alcune proteine, ma la stragrande maggioranza (oltre il 95%) è inutilizzata e viene metabolizzato attraverso un gruppo di enzimi che alla fine generano acetoacetato e malato.Nell’alcaptonuria, l’enzima HGD non è in grado di metabolizzare l’acido omogentisico (generato dalla tirosina) in 4-maleilacetoacetato de i livelli di acido omogentisico nel sangue sono 100 volte più alti di quanto ci si aspetterebbe normalmente, nonostante il fatto che una quantità sostanziale venga eliminata nel urina dai reni. L’acido omogentisico viene convertito nella sostanza correlata acido benzochinone acetico che forma polimeri che assomigliano al pigmento della pelle melanina. Questi si depositano nel collagene, una proteina del tessuto connettivo, di particolari tessuti come la cartilagine. Questo processo è chiamato ocronosi (poiché il tessuto appare ocra); il tessuto ocronotico è irrigidito e insolitamente fragile, compromettendo la sua normale funzione e causando danni.
Trasmissione
L’alcaptonuria è una malattia genetica recessiva. Una malattia è detta a trasmissione autosomica recessiva quando l’allele alterato deve essere presente in coppia (omozigosi), cioè sono necessarie due copie dell’allele difettoso per far sì che la malattia si esprima, a prescindere dal sesso. Non basta un solo genitore portatore sano o malato, bensì entrambi i genitori devono essere portatori sani o malati. Il fenotipo quindi si esprime quando nel genotipo dell’individuo sono presenti entrambi gli alleli responsabili, fatto che spiega l’alta probabilità di sviluppare malattie genetiche in caso di incesto. Quindi:
- un individuo che possegga entrambi gli alleli alterati: è portatore ed è malato;
- un individuo che possegga solo un allele alterato: è portatore ma è sano;
- un individuo che non possegga nessun allele alterato: NON è portatore ed è sano.
Essere portatore sano vuol dire quindi NON avere la patologia ma possedere nel proprio genotipo un allele mutato, che può essere trasmesso alle generazioni successive.
Dalla combinazione delle possibili condizioni di genitori sani, malati e portatori sani, deriva la distribuzione probabilità che la malattia sia trasmessa ai figli:
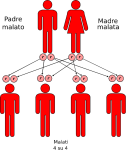
- genitori malato-malato: la probabilità che il figlio/a nasca malato è del 100%;
- genitori sano-malato: la probabilità che il figlio/a nasca portatore sano è del 100%;
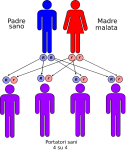
- genitori malato-portatore sano: la probabilità che il figlio/a nasca malato è del 50% e del 50% che nasca portatore sano;
- genitori sano-portatore sano: la probabilità che il figlio/a nasca sano è del 50% e del 50% che nasca portatore sano;
- genitori portatore-portatore: la probabilità che il figlio/a nasca portatore sano è del 50% mentre è del 25% che nasca sano o malato.
Se nessuno dei genitori ha un allele mutato, non c’è ovviamente alcuna trasmissione autosomica recessiva ed i figli saranno tutti sani e NON portatori dell’allele mutato.
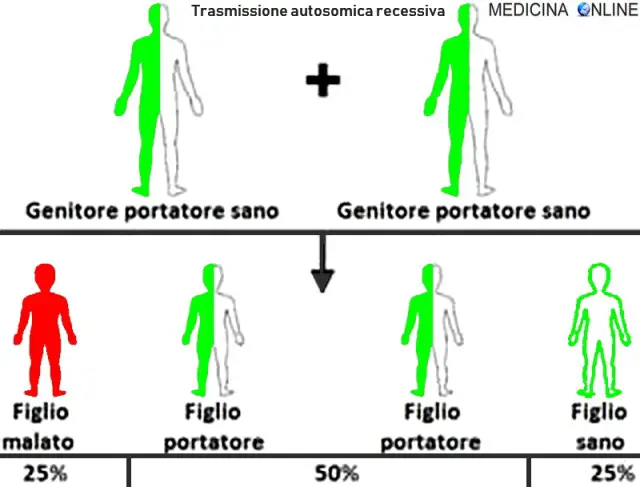
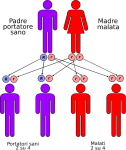
Nell’immagine che segue, è raffigurata la tipica situazione in cui entrambi i genitori sono sani ma portatori dell’allele mutato:
- un figlio su quattro avrà entrambi gli alleli alterati e sarà malato ed ovviamente portatore;
- due figli su quattro avranno un allele normale ed uno alterato e saranno sani ma anche portatori;
- un figlio su quattro avrà entrambi gli alleli normali e sarà sano e NON portatore.
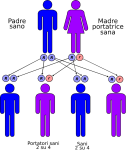
Le altre quattro situazioni possibili sono raffigurate nelle seguenti immagini:
Segni e sintomi
I pazienti con alcaptonuria sono asintomatici in genere da bambini o in età giovane adulta, tuttavia la loro urina può diventare marrone o addirittura nera come l’inchiostro se raccolta e lasciata esposta all’aria aperta. La pigmentazione può essere notata nella cartilagine dell’orecchio e in altra cartilagine e nella sclera e nel limbus corneale dell’occhio. Dopo i 30 anni o – in alcuni casi – dopo i 40 anni, le persone iniziano a sviluppare dolore alle articolazioni portanti della colonna vertebrale, delle anche e delle ginocchia. Il dolore può essere grave al punto da interferire con le attività della vita quotidiana e può influire sulla capacità di muoversi e di lavorare: ciò abbassa fortemente la qualità della vita del paziente, con conseguenze psicologiche anche gravi (depressione e, nei casi più gravi, ideazioni suicidari). La chirurgia di sostituzione dell’articolazione (anca e spalla) è spesso necessaria in età relativamente giovane. A lungo termine, il coinvolgimento delle articolazioni spinali porta a un movimento ridotto della gabbia toracica e può influire sulla respirazione: possibile quindi una dispnea. La densità minerale ossea può essere influenzata, aumentando il rischio di osteoporosi e fratture ossee; più frequente anche la rottura di tendini e muscoli. Possono verificarsi cardiopatie valvolari, principalmente calcificazione e rigurgito delle valvole aortica e mitrale e, nei casi gravi e progressivi, può essere necessaria la sostituzione della valvola. Le irregolarità del ritmo cardiaco e l’insufficienza cardiaca colpiscono una percentuale significativa di persone con alcaptonuria (40% e 10%, rispettivamente). La perdita dell’udito colpisce il 40% delle persone. Inoltre, esiste una propensione allo sviluppo di calcoli renali e di calcoli in altre parti del corpo. Possono verificarsi anche calcoli biliari e calcoli nella prostata e nelle ghiandole salivari (scialolitiasi).
Diagnosi
Se si sospetta la diagnosi di alcaptonuria, questa può essere confermata o esclusa raccogliendo l’urina delle 24 ore o un campione di sangue, determinando la quantità di acido omogentisico mediante cromatografia su carta o cromatografia su strato sottile, utilizzando egualmente il sangue o l’urina. Nei soggetti sani l’acido omogentisico è assente, mentre nei soggetti affetti i livelli plasmatici risultano intorno a 6.6 µg/ml e i livelli urinari risultano mediamente 3.12 mmol per ogni mmol di creatinina. E’ disponibile il test genetico per l’alcaptonuria. La gravità dei sintomi e la risposta al trattamento possono essere quantificate attraverso un questionario validato intitolato AKU Severity Score Index. Questo assegna punteggi alla presenza di particolari sintomi e caratteristiche, come la presenza di pigmentazione degli occhi e della pelle, dolori articolari, problemi cardiaci e calcoli d’organo.
Trattamento
Nessuna modalità di trattamento è stata inequivocabilmente dimostrata per ridurre le complicanze dell’alcaptonuria. I principali tentativi di trattamento si sono concentrati sulla prevenzione dell’ocronosi attraverso la riduzione dell’accumulo di acido omogentisico. Tali trattamenti comunemente raccomandati includono grandi dosi di acido ascorbico (vitamina C) e la restrizione dietetica degli amminoacidi fenilalanina e tirosina. Il trattamento con vitamina C, tuttavia, non ha un’efficacia definitivamente dimostrata e la restrizione proteica (che può essere difficile da rispettare) non si è dimostrata sempre efficace.
Leggi anche: Dieta ipoproteica ed aproteica: cosa mangiare e chi la deve seguire
Nitisinone
Diversi studi hanno suggerito che il nitisinone (un erbicida) può essere efficace nel trattamento dell’alcaptonuria. Il nitisinone inibisce l’enzima 4-idrossifenilpiruvato diossigenasi, responsabile della conversione della tirosina in acido omogentisico, bloccando così la produzione e l’accumulo di HGA. Il nitisinone è stato utilizzato per qualche tempo a dosi molto più elevate nel trattamento della tirosinemia di tipo I. È stato dimostrato che il trattamento con nitisinone provoca una riduzione superiore al 95% dell’HGA plasmatico e urinario. Lo svantaggio principale è l’accumulo di tirosina, i cui rischi a lungo termine sono sconosciuti; esiste una preoccupazione particolare per i danni alla cornea dell’occhio. L’uso a lungo termine richiede un monitoraggio frequente per le complicanze. Nel 2020 l’Agenzia europea per i medicinali ha approvato il farmaco Orfadin (nitisinone) per il trattamento dell’alcaptonuria nei pazienti adulti
Prognosi
L’alcaptonuria non sembra influenzare l’aspettativa di vita, anche se l’ultimo studio sull’argomento è del 1985. L’impatto principale è sulla qualità della vita del paziente: molte persone con alcaptonuria hanno infatti sintomi cronicamente invalidanti come dolore, disturbi del sonno e sintomi respiratori. Questi sintomi generalmente iniziano nella terza o quarta decade di vita. L’età tipica in cui è necessario un intervento di sostituzione articolare è di 50-55 anni.
Leggi anche:
- Ocronosi endogena ed esogena: cause, trasmissione, sintomi, diagnosi e terapie
- Tirosinemia: cause, trasmissione, sintomi, diagnosi e terapie
- Fenilchetonuria: sintomi, valori, alimentazione, diagnosi
- Mioglobinuria genetica ricorrente: cause, sintomi, diagnosi
- Mioglobinuria (mioglobina nelle urine): significato, cause, esame delle urine, cure
- Esami per valutare funzionalità renale ed insufficienza renale
- Esame delle urine completo con urinocoltura: come farlo e capire i risultati
- Emoglobina nelle urine (emoglobinuria): cause, sintomi e terapia
- Ematuria da sforzo e da esercizio fisico: cause e terapia
- Proteinuria 24 ore alta: cause, tipi, valori e terapie
- Batteriuria: asintomatica, non significativa, valori normali, cura
- Scura o chiara, liquida o schiumosa: la tua urina rivela la tua salute
- Insufficienza renale cronica: stadi, dieta, sintomi, diagnosi e terapia
- Iperuricemia, ipericosuria, acido urico nel sangue e nelle urine
- Differenza tra emoglobina e mioglobina
- Differenza tra ematuria iniziale, terminale e totale ed ipotesi diagnostiche
- Emodialisi: come funziona, effetti collaterali e complicanze
- Quali sono i sintomi di malattia o cattivo funzionamento dei reni?
- Differenza tra esame delle urine ed urinocoltura
- Mi alzo spesso di notte per urinare: quali sono le cause e le cure?
- CPK (CK) alto o basso: cause, conseguenze, valori, cure, cosa indica
- Miastenia gravis e malattia del timo: cause, sintomi, diagnosi, terapia
- Le tue feci dicono se sei in salute: con la Scala di Bristol impara ad interpretarle
- Emoglobina bassa, alta, cause e valori normali
- Differenza tra sindrome nefritica e nefrosica
- Differenza tra proteinuria transitoria, persistente ed ortostatica
- Uroflussometria: indicazioni, preparazione, come si esegue
- Vescica: dove si trova, anatomia, funzioni e patologie frequenti in sintesi
- Rene: anatomia, funzioni e patologie in sintesi
- Azotemia (Urea) alta o bassa: valori, cause, sintomi e cosa fare
- Tumore alla vescica: terapie, asportazione, si può guarire?
- Bruciore e stimoli frequenti di urinare: cistite, sintomi e cure
- Perché viene la cistite e come curarla?
- Differenza tra anuria e ritenzione urinaria
- Esame delle urine completo con urinocoltura: come fare e capire i risultati
- Urodinamica: cos’è, a che serve e come funziona
- Minzione: come funziona l’emissione di urina e come si controlla
- Uretra maschile e femminile: anatomia, funzioni e patologie in sintesi
- Uretere: dove si trova, anatomia, funzioni e patologie in sintesi
- Fa male trattenere l’urina troppo a lungo? Per quale motivo?
- Differenze tra apparato urinario maschile e femminile
- Quante volte al giorno è normale urinare? Vescica iperattiva e ansia
- Prostata: anatomia, dimensioni, posizione e funzioni in sintesi
- Ecografia prostatica transrettale: come si svolge, è dolorosa, a che serve?
- PSA totale e free alto: capire i risultati dell’esame e rischio di tumore alla prostata
- Esplorazione rettale digitale della prostata: fa male? A che serve?
- Prostata ingrossata ed infiammata: ecco cosa fare per mantenerla in salute
- Vescica neurogena disinibita, riflessa, autonoma, atonica
- Albumina ed albuminemia alta o bassa: cause, valori e terapie
- Parassiti e vermi nelle feci: sintomi e come eliminarli con farmaci e rimedi naturali
- Coprocoltura e antibiogramma: procedura e perché si eseguono
- Esame e raccolta delle feci: come si fa nel modo corretto ed a che serve
- Differenza tra globuli rossi, bianchi e piastrine
- Feci con sangue, muco, cibo: quando preoccuparsi?
- Emocromo: guida completa a tutti i valori del sangue normali e patologici
- Emocromo: valori di riferimento e significato clinico [SCHEMA]
- Ematocrito (HCT): basso, alto, in gravidanza, valori normali e interpretazione
- Indici corpuscolari MCV, MCH, MCHC, RDW: cosa sono ed a che servono
- Volume corpuscolare medio (MCV): alto, basso, valori normali e significato
- MCH alto, basso, valori normali ed interpretazione
- MCHC alto, basso, valori normali ed interpretazione
- RDW alto, basso, valori normali ed interpretazione
- Ormoni tiroidei: differenza T3 e T4, valori normali e patologici
- TSH alto, basso e valori normali: qual è il significato clinico?
- Tireoglobulina alta, bassa, valori normali ed interpretazione
- Globuli rossi (eritrociti) alti, bassi, valori normali e interpretazione
- Globuli bianchi (leucociti) alti, bassi, valori normali ed interpretazione
- Eosinofili alti, bassi, valori normali ed interpretazione
- Neutrofili alti, bassi, valori normali ed interpretazione
- Basofili alti, bassi, valori normali ed interpretazione
- Differenza tra anemia ed anemia mediterranea (talassemia)
- Differenza tra anemia e leucemia
- Perché la mononucleosi è chiamata anche “malattia del bacio”?
- Leucemia: sintomi, cause, cure e le diverse forme
- Leucemia mieloide acuta: cause, sintomi, diagnosi e cura
- Mieloma multiplo: cause, sintomi, diagnosi e cura
- Microcitemia (talassemia) : cause, sintomi, diagnosi e cura
- Differenza tra anemia e microcitemia
- Differenza tra anemia megaloblastica e perniciosa
- Differenza tra anemia mediterranea e falciforme
- Differenza tra emoglobina fetale ed adulta
- Differenza tra emoglobina, ferro, ferritina e transferrina
- Differenza tra emoglobina e globuli rossi
- Differenza tra emocromo ed ematocrito
- Surrene: anatomia, funzioni e patologie in sintesi
- Differenza tra renella e calcoli renali
- Rene: anatomia, funzioni e patologie in sintesi
- Differenza tra surrene e rene
- Differenza tra rene policistico e multicistico
- Differenza tra rene destro e sinistro
- Differenza tra nefrologo ed urologo: patologie e competenze specifiche e comuni
- Testosterone basso, alto, valori normali ed interpretazione
- Ormone follicolo stimolante (FSH) alto, basso, valori normali e significato
- Lingua bianca, impastata, spaccata: cause e quando è pericolosa
- Lingua bianca, impastata, spaccata: cure e rimedi naturali
- Smegma femminile, vulviti e cattivi odori vaginali: cura e prevenzione
- Mi dicevano “sei grassa” così decisi che non avrei mangiato più. Mai più. La testimonianza di una paziente anoressica
- Anoressia: le immagini drammatiche di un corpo che non esiste più
- Si mette a dieta e perde 60 kg. La ragione per cui lo fa vi lascerà senza parole
- Apparato urinario: anatomia e fisiologia [SCHEMA]
- Filtrazione glomerulare, riassorbimento e secrezione
- Differenza tra arteriola afferente ed efferente: struttura e funzioni
- Glomerulo renale: schema, funzione e flusso ematico renale
- Qual è la differenza tra arteria e vena?
- Trombo: cause, classificazione, trombosi venose, arteriose e sistemiche
- Differenza tra trombosi arteriosa e venosa profonda e superficiale
- Differenza tra trombo e placca aterosclerotica
- Differenza tra pressione arteriosa e venosa
- Differenza tra pressione massima (sistolica), minima (diastolica) e differenziale
- Pressione arteriosa: valori normali e patologici
- Pressione alta (ipertensione arteriosa): sintomi, cause, valori e cure
- Perché la pressione arteriosa alta (ipertensione) è pericolosa?
- Ipertermia maligna: cause, fattori di rischio, sintomi, rischi, cure, mortalità
- Rabdomiolisi: sintomi, da statine, conseguenze, valori
- Ustione di terzo grado: cosa fare e tempi di guarigione della pelle
Dott. Emilio Alessio Loiacono
Medico Chirurgo
Direttore dello Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o unisciti al nostro gruppo Facebook o ancora seguici su Twitter, su Instagram, su Tumblr e su Pinterest, grazie!
