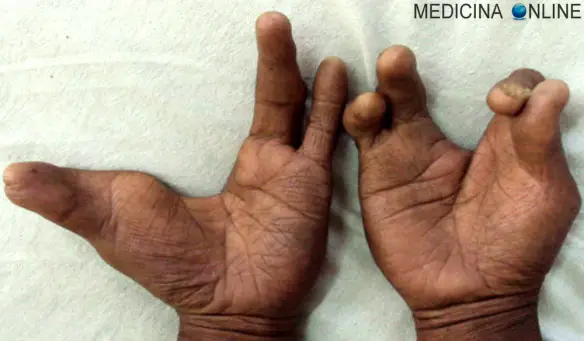
La polidattilia (anche chiamata iperdattilia) è un’anomalia genetica caratterizzata da dita in soprannumero nelle mani o nei piedi. Rispetto alla normale conformazione possono essere presenti uno, due o anche più dita per ogni estremità. La polidattilia è un carattere a trasmissione autosomica dominante, a penetranza incompleta (cioè non tutti i portatori del gene alterato esprimono questa malattia) e con un genotipo che non sempre corrisponde a medesimo fenotipo (due persone con identica genetica, possono sviluppare una malformazione diversa). La polidattilia può presentarsi da sola, senza altre patologie o malformazioni concomitanti (“polidattilia NON sindromica“), o più spesso come una delle caratteristiche di una data sindrome ed in questo caso prende il nome di “polidattilia sindromica“. La polidattilia può associarsi anche ad altri difetti malformativi della mano, ad esempio la signora nella foto in alto ha – nella mano sinistra – un dito in soprannumero fuso con il pollice, malformazione chiamata sindattilia.
La polidattilia (anche chiamata iperdattilia) è un’anomalia genetica caratterizzata da dita in soprannumero nelle mani o nei piedi. Rispetto alla normale conformazione possono essere presenti uno, due o anche più dita per ogni estremità. La polidattilia è un carattere a trasmissione autosomica dominante, a penetranza incompleta (cioè non tutti i portatori del gene alterato esprimono questa malattia) e con un genotipo che non sempre corrisponde a medesimo fenotipo (due persone con identica genetica, possono sviluppare una malformazione diversa). La polidattilia può presentarsi da sola, senza altre patologie o malformazioni concomitanti (“polidattilia NON sindromica“), o più spesso come una delle caratteristiche di una data sindrome ed in questo caso prende il nome di “polidattilia sindromica“. La polidattilia può associarsi anche ad altri difetti malformativi della mano, ad esempio la signora nella foto in alto ha – nella mano sinistra – un dito in soprannumero fuso con il pollice, malformazione chiamata sindattilia.
Caratteristiche del dito in soprannumero
Il dito soprannumerario è solitamente un piccolo pezzo di tessuto molle; occasionalmente contiene ossa prive di giunture. Spesso è non funzionante, mentre è raro che sia completo e funzionante. La polidattilia può essere di tre tipi principali:
- polidattilia postassiale: il dito supplementare è presente nel lato della mano dal lato del mignolo (più frequente);
- polidattilia preassiale: il dito supplementare è presente nel lato della mano dal lato del pollice (meno frequente);
- polidattilia centrale: il dito supplementare è presente tra le dita centrali (indice, medio, anulare) della mano (più raro).
Diagnosi della polidattilia
La diagnosi della polidattilia è clinica, ma si avvale anche di esami di diagnostica per immagini, tipicamente una radiografia della mano. Di solito, ai portatori di questa malattia viene richiesto anche un test genetico per verificare possibili anomalie genetiche. Grazie al test, i medici sono in grado di valutare se la patologia è sindromica oppure no.
Malformazioni sindromiche della mano
Le malformazioni congenite della mano su base genetica vengono classificate in 3 gruppi che includono difetti in riduzione, difetti della differenziazione e difetti in eccesso (polidattilie). Un esempio di difetto in riduzione (dita in numero minore) è la sindrome di Holt-Oram la quale è una condizione a trasmissione dominante caratterizzata da anomalie del pollice, che può essere ipoplastico, assente o trifalangeo, e difetti cardiaci. Il gene responsabile di questa condizione è TBX5, che appartiene ad una famiglia di geni che codificano fattori di trascrizione. Appartengono ai difetti della differenziazione le brachidattilie e le sindattilie. Le sindattilie non sindromiche sono di 5 tipi, tutti a trasmissione autosomica dominante. La sindattilia tipo 2 conosciuta anche come sinpolidattilia interessa il 3° e 4° dito, e si associa a polidattilia del 4° dito. Si tratta quindi di una forma mista dovuta a mutazione del gene HOXD13.
Leggi anche:
Polidattilie sindromiche e terapie
L’esatto meccanismo molecolare responsabile delle polidattilie non è stato completamente chiarito. Tuttavia esse sono state associate a 39 mutazioni genetiche. Un esempio è dato dalla polidattilia pre-assiale tipo IV causata da mutazioni del gene GLI3. In caso di polidattilia sindromica, oltre al dito soprannumerario, sono presenti nel paziente una serie di altri sintomi di varia natura, spesso altre marformazioni osteo-articolari.
Trattamento della polidattilia
Riguardo al trattamento, la nascita di un bambino con un difetto congenito degli arti impone un approccio multidisciplinare che coinvolge diverse figure professionali al fine di ottenere i migliori risultati. Quando il difetto è isolato oppure fa parte di un quadro sindromico che non è letale, accanto alla valutazione del neonatologo e del genetista clinico che concorrono a formulare una diagnosi, è necessario l’intervento di altri specialisti come il chirurgo plastico, l’ortopedico, il fisiatra e altri a seconda del quadro malformativo osservato.
Polidattilia: quando fare l’interento chirurgico?
Fino ad alcuni anni fa si interveniva dopo il secondo anno di vita, ma oggi, grazie all’avanzamento delle tecniche chirurgiche, si preferisce effettuare l’intervento fra i 6 e 18 mesi di età.
Leggi anche:
- Dita ippocratiche congenite e secondarie: cause, sintomi e terapie
- Quante ossa ci sono nella mano e come si chiamano?
- Sindrome di Marfan: trasmissione, sintomi e frequenza
- Le malattie genetiche più diffuse al mondo
- Donne nate senza vagina: è la sindrome di Rokitansky-Kuster-Hauser
- Sindrome di Klinefelter: cariotipo, cause, sintomi e cura
- Microtia ed anotia: la malformazione congenita dell’orecchio
- Palatoschisi: cause, problemi derivanti e cure
- Labbro leporino (cheiloschisi): tipi, cause, problematiche e cure
- Arinia: la mancanza congenita del naso
- Sindrome di Turner: cariotipo, cause, sintomi e segni caratteristici
- Sindrome di Down: cause, sintomi in gravidanza e nei neonati
- Atrofia muscolare progressiva: cause, sintomi, cura, aspettativa di vita
- Differenze tra sclerosi laterale amiotrofica e sclerosi multipla
- Morbo di Parkinson: cause, sintomi, decorso, terapie
- Morbo di Alzheimer: cause, sintomi, decorso, terapie
- Demenza senile: cause, sintomi, decorso e cure
- Sindrome del cuore infranto: il falso infarto di chi ha il “cuore spezzato”
- Sindrome di Cornelia de Lange: avere l’aspetto di un bambino per tutta la vita
- Sindrome di Wolff-Parkinson-White: cos’è, cosa fare, come si cura
- Quando il paziente deceduto “resuscita”: la Sindrome di Lazzaro
- La sindrome che fa crescere il pene alle bambine di 12 anni
- Fibrosi cistica polmonare: cos’è, sintomi in neonati e bambini, cure
- La Sindrome da abbandono: cos’è e come si supera
- Malattia di Huntington: cos’è, ereditarietà, come si trasmette, età di insorgenza
- Anemia falciforme: cosa significa, cause, sintomi e cure
- Differenze tra la distrofia muscolare di Duchenne e di Becker
- Talassemia: cos’è, sintomi, cure, differenti tipi ed alimentazione
- Celiachia: cos’è il glutine, in quali alimenti è contenuto ed in quali no?
- Sindrome di Noonan: cause, sintomi nel neonato, aspettative di vita
- Sindrome di Bloom: cause, sintomi, diagnosi e terapia
- Insensibilità congenita al dolore: la strana malattia che non ti fa sentire nessun dolore
- Cos’è un cromosoma ed a che serve?
- Differenza tra gene e allele
- Quanti cromosomi hanno esseri umani, scimmie, cani, gatti e topi?
- Quanti cromosomi ha chi è affetto da Sindrome di Down?
Lo staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o seguici su Twitter, su Instagram o su Pinterest, grazie!
Condividi questo articolo: