
 La sindrome di Gilbert è una patologia benigna del fegato che si manifesta con iperbilirubinemia, cioè un aumento della bilirubina nel sangue, spesso nel Continua a leggere
La sindrome di Gilbert è una patologia benigna del fegato che si manifesta con iperbilirubinemia, cioè un aumento della bilirubina nel sangue, spesso nel Continua a leggere
Archivi tag: malattia genetica
Fenilchetonuria: sintomi, valori, alimentazione, diagnosi
 Con “fenilchetonuria” o “iperfenilalaninemia di tipo I” in medicina si indica la presenza di elevate concentrazione di Continua a leggere
Con “fenilchetonuria” o “iperfenilalaninemia di tipo I” in medicina si indica la presenza di elevate concentrazione di Continua a leggere
Differenza genetica tra uomo e scimmia
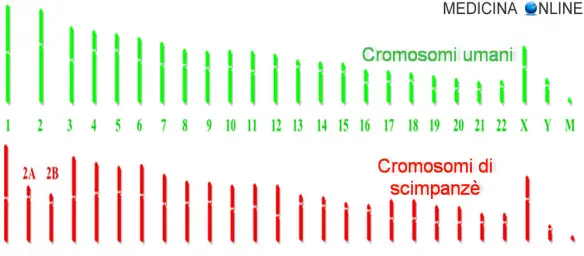
Confronto tra il genoma umano e dello scimpanzé. “M” indica il DNA mitocondriale
Confrontando con metodi computerizzati (denominati sequenziamento del DNA) il genoma umano (Homo sapiens) con quello dello scimpanzé comune (Pan troglodytes) si constata che le due specie hanno circa il 99% dei geni in comune (ossia che su 3 miliardi di basi, pari a 1 miliardo di codoni, soltanto 30 milioni di basi sono diverse).
Cromosomi di uomo e scimpanzè
I cromosomi dell’essere umano e dello scimpanzé sono decisamente simili, molto più di quanto si potrebbe immaginare. La principale differenza è che gli umani hanno un paio Continua a leggere
Differenza tra genetica e genomica
Rispondi
 Genetica
Genetica
La genetica è la branca della biologia che studia i geni, l’ereditarietà e la variabilità genetica negli organismi viventi, focalizzandosi sulla comprensione dei meccanismi alla base di questi fenomeni, noti sin dall’antichità ma spiegati solo nella metà del 1800 dal biologo ceco Gregor Mendel.
Genomica
La genomica è una branca della genetica che si occupa in particolare dello studio di struttura, contenuto, funzione ed evoluzione del genoma degli organismi viventi, specie Continua a leggere
Differenza tra genetica ed epigenetica
 Genetica
Genetica
La genetica è la branca della biologia che studia i geni, l’ereditarietà e la variabilità genetica negli organismi viventi, focalizzandosi sulla comprensione dei meccanismi alla base di questi fenomeni, noti sin dall’antichità ma spiegati solo nella metà del 1800 dal biologo ceco Gregor Mendel. I “caratteri” mendeliani dell’individuo, ricevuti dai propri genitori, corrispondono a sequenze di DNA (acido desossiribonucleico) chiamate geni e Continua a leggere
Differenza tra malattia genetica, ereditaria e congenita
 Che differenza c’è tra malattia genetica, ereditaria e congenita? Questi termini vengono spesso confusi o usati come sinonimi, sbagliando.
Che differenza c’è tra malattia genetica, ereditaria e congenita? Questi termini vengono spesso confusi o usati come sinonimi, sbagliando.
Malattia congenita
Per cominciare si definisce malattia congenita un qualsiasi tipo di patologia presente nell’individuo sin dalla sua nascita. Una malattia congenita può essere genetica o meno: ad esempio una malformazione dovuta a eventi verificatisi durante la gravidanza non è genetica. Una malattia congenita non è quindi trasmessa necessariamente dai Continua a leggere
Aracnodattilia, segno del pollice e del polso, Sindrome di Marfan e di Beals
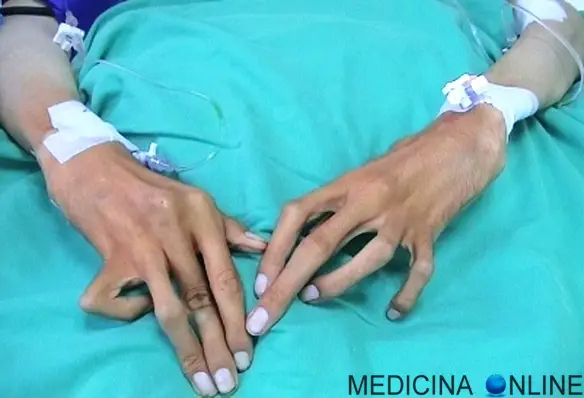 Con “aracnodattilia” (“dita a ragno”) si intende una deformità dello scheletro delle mani caratterizzata da dita affusolate e sproporzionatamente allungate rispetto al palmo della mano o alla pianta del piede. Queste estremità possono risultare, inoltre, insolitamente flessibili, similmente alle zampe di un ragno, motivo per cui questa patologia è così chiamata. Costituisce uno dei sintomi della Sindrome di Marfan ed è caratterizzata dal “segno del pollice” e dal “segno del polso”. L’aracnodattilia può essere congenita e svilupparsi nel contesto di diverse malattie genetiche. Altre persone possono manifestare questo allungamento patologico delle dita delle mani e dei piedi nel tempo, durante l’infanzia o l’età adulta.
Con “aracnodattilia” (“dita a ragno”) si intende una deformità dello scheletro delle mani caratterizzata da dita affusolate e sproporzionatamente allungate rispetto al palmo della mano o alla pianta del piede. Queste estremità possono risultare, inoltre, insolitamente flessibili, similmente alle zampe di un ragno, motivo per cui questa patologia è così chiamata. Costituisce uno dei sintomi della Sindrome di Marfan ed è caratterizzata dal “segno del pollice” e dal “segno del polso”. L’aracnodattilia può essere congenita e svilupparsi nel contesto di diverse malattie genetiche. Altre persone possono manifestare questo allungamento patologico delle dita delle mani e dei piedi nel tempo, durante l’infanzia o l’età adulta.

Cos’è il “segno del pollice”?
L’aracnodattilia si riconosce per la positività del “segno del pollice”, che consiste nel far chiudere il pugno con all’interno il pollice: se si osserva la protrusione della falange distale dello stesso dito, oltre il bordo ulnare, allora il segno del pollice è detto “positivo” ed indica la presenza di aracnodattilia (vedi foto in alto a sinistra).
Cos’è il “segno del polso”?
Altro esame utile nella valutazione della patologia in questione, è il “segno del polso”: se le falangi distali di pollice e mignolo si sovrappongono, quando poste a cingere il polso, il segno del polso è “positivo” ed indica aracnodattilia (vedi foto in alto a destra).
Patologie in cui è presente l’aracnodattilia
L’aracnodattilia costituisce tipicamente un sintomo della Sindrome di Marfan, dipendendo da un’alterazione nel metabolismo del collagene. Questa deformità rientra però anche tra le anomalie che si possono riscontrare nell’omocisteinuria e nella Sindrome di Beals (malattia del tessuto connettivo che si presenta in modo simile alla Sindrome di Marfan). In qualche caso, però, l’aracnodattilia può presentarsi in assenza di una malattia di base.
Leggi anche:
- Sindrome di Marfan: trasmissione, sintomi e frequenza
- Ectrodattilia: cause, cure ed immagini
- Polidattilia; cause, ereditarietà, sindromica e chirurgia
- Sindrome di Beals e padiglione auricolare “accartocciato”: sintomi e cure
- Brachidattilia a mano o piede: trasmissione ereditaria e cura
- Differenza tra polidattilia, sindattilia, ectrodattilia, oligodattilia e aracnodattilia
- Dita ippocratiche congenite e secondarie: cause, sintomi e terapie
- Le malattie genetiche più diffuse al mondo
- Quante ossa ci sono nella mano e come si chiamano?
- Donne nate senza vagina: è la sindrome di Rokitansky-Kuster-Hauser
- Microtia ed anotia: la malformazione congenita dell’orecchio
- Palatoschisi: cause, problemi derivanti e cure
- Labbro leporino (cheiloschisi): tipi, cause, problematiche e cure
- Arinia: la mancanza congenita del naso
- Sindrome di Klinefelter: cariotipo, cause, sintomi e cura
- Sindrome di Turner: cariotipo, cause, sintomi e segni caratteristici
- Sindrome di Down: cause, sintomi in gravidanza e nei neonati
- Atrofia muscolare progressiva: cause, sintomi, cura, aspettativa di vita
- Differenze tra sclerosi laterale amiotrofica e sclerosi multipla
- Morbo di Parkinson: cause, sintomi, decorso, terapie
- Morbo di Alzheimer: cause, sintomi, decorso, terapie
- Demenza senile: cause, sintomi, decorso e cure
- Sindrome del cuore infranto: il falso infarto di chi ha il “cuore spezzato”
- Sindrome di Cornelia de Lange: avere l’aspetto di un bambino per tutta la vita
- Sindrome di Wolff-Parkinson-White: cos’è, cosa fare, come si cura
- Quando il paziente deceduto “resuscita”: la Sindrome di Lazzaro
- La sindrome che fa crescere il pene alle bambine di 12 anni
- Fibrosi cistica polmonare: cos’è, sintomi in neonati e bambini, cure
- La Sindrome da abbandono: cos’è e come si supera
- Malattia di Huntington: cos’è, ereditarietà, come si trasmette, età di insorgenza
- Anemia falciforme: cosa significa, cause, sintomi e cure
- Differenze tra la distrofia muscolare di Duchenne e di Becker
- Talassemia: cos’è, sintomi, cure, differenti tipi ed alimentazione
- Celiachia: cos’è il glutine, in quali alimenti è contenuto ed in quali no?
- Sindrome di Noonan: cause, sintomi nel neonato, aspettative di vita
- Sindrome di Bloom: cause, sintomi, diagnosi e terapia
- Insensibilità congenita al dolore: la strana malattia che non ti fa sentire nessun dolore
- Cos’è un cromosoma ed a che serve?
- Differenza tra gene e allele
- Quanti cromosomi hanno esseri umani, scimmie, cani, gatti e topi?
- Quanti cromosomi ha chi è affetto da Sindrome di Down?
Lo staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o seguici su Twitter, su Instagram o su Pinterest, grazie!
Sindrome di Turner: cariotipo, cause, sintomi e segni caratteristici
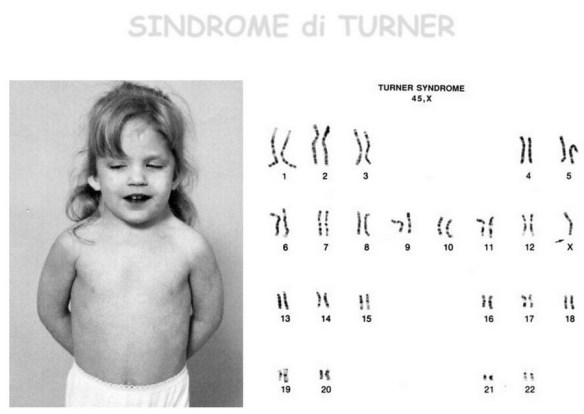 La sindrome di Turner è una sindrome cromosomica caratterizzata da bassa statura, disgenesia gonadica in assenza di ambiguità dei genitali (difetti dello sviluppo dei caratteri sessuali secondari ed infertilità), segni caratteristici del fenotipo esterno ed anomalie di alcuni organi interni.
La sindrome di Turner è una sindrome cromosomica caratterizzata da bassa statura, disgenesia gonadica in assenza di ambiguità dei genitali (difetti dello sviluppo dei caratteri sessuali secondari ed infertilità), segni caratteristici del fenotipo esterno ed anomalie di alcuni organi interni.
Epidemiologia
La prevalenza è di 1/2000-1/2500 nate di sesso femminile.
Eziologia
La sindrome di Turner è causata dall’assenza parziale o totale di uno dei due cromosomi sessuali X, con o senza linee cellulari a mosaico. Tale evento sembra essere casuale e non legato a condizioni predisponenti materne o paterne. Il cariotipo è nel 30-40% dei casi 45,X (monosomia della X completa ed omogenea), nel 60-70% dei casi a mosaico (con una linea cellulare 45,X e una linea cellulare normale 46,XX) o con anomalie strutturali di un cromosoma X: isocromosoma [X i(Xq)], ad anello [r(X)], delezione del braccio lungo o del braccio corto della X [del(X)]. Si ritiene che circa il 10% degli aborti spontanei abbia un cariotipo 45,X.
Segni e sintomi
I soggetti affetti presentano un fenotipo femminile, ma nell’85-90% dei casi, a causa della disgenesia gonadica, non sviluppano o sviluppano solo parzialmente i caratteri sessuali secondari oppure vanno incontro a menopausa precoce. I segni fenotipici esterni caratteristici, definiti turneriani, sono espressi in modo estremamente variabile. Nella maggior parte dei casi sono molto sfumati e riconoscibili solo da parte di esperti. I segni tipici interessano in particolare il viso (presenza di ptosi palpebrale ed epicanto), le orecchie (spesso grandi e basso-impiantate), il collo (spesso corto con cute abbondante), il torace (largo con capezzoli distanziati) e lo scheletro (cubito valgo, segno di Madelung a livello dell’avambraccio, frequente scoliosi in epoca puberale, accorciamento del IV e V dito della mano o/e del piede). L’età ossea è generalmente ritardata rispetto all’età cronologica e la sua lenta progressione è secondaria all’assenza di un’ adeguata produzione di ormoni sessuali.
- Apparato cardiovascolare. Le malformazioni degli organi interni interessano in particolare l’apparato cardiovascolare. La sindrome di Turner si associa ad una prevalenza del 25% circa di malformazioni cardiovascolari, rispetto al 2% della popolazione generale. Le cardiopatie congenite interessano prevalentemente il cono di efflusso del cuore sinistro: valvola aortica bicuspide, coartazione aortica, valvulopatia aortica, ma anche il cuore destro (ritorno venoso anomalo parziale delle vene polmonari). Nel periodo prenatale possono essere individuati ecograficamente segni caratteristici della sindrome di Turner, che correlano con un aumentato rischio di cardiopatia congenita: aumentata translucenza nucale, igroma cistico ed idrope fetale. Le cardiopatie congenite vengono oggi trattate con successo precocemente mediante correzione chirurgica. Recenti studi hanno evidenziato in questi soggetti affette da sindrome di Turner anche aumentata incidenza di dilatazione dell’aorta, a livello della radice e dell’aorta ascendente, anche in soggetti senza fattori predisponenti, quali la presenza di cardiopatia congenita od ipertensione. E’ stata inoltre riportata una precoce compromissione della funzionalità endoteliale vascolare ed ipotizzato un difetto della parete arteriosa, correlato probabilmente a fattori genetici ed alla carenza di estrogeni. L’ipertensione arteriosa si manifesta precocemente e circa il 50% delle adulte hanno ipertensione clinica con profilo pressorio notturno anormale.
- Apparato urinario. Le anomalie dell’apparato urinario si riscontrano con maggiore frequenza rispetto alla popolazione generale (30% vs 3.2%); il rene a ferro di cavallo è la malformazione piu’ frequente, ma generalmente non comporta anomalie funzionali. Nel 15% dei casi sono presenti anomalie della pelvi o degli ureteri evidenziabili peraltro in epoca prenatale che, in alcuni casi (idronefrosi), necessitano di un trattamento chirurgico
- Orecchio. I soggetti con sindrome di Turner hanno piu’ elevata prevalenza di patologia dell’orecchio medio e, in età adulta, dell’orecchio interno. Molto importante è il follow-up ORL per la ricorrenza di otiti, che vanno prevenute e trattate opportunamente per evitare sequele audiologiche.
- Autoimmunità. Frequenti sono le malattie autoimmuni, in particolare le tiroiditi autoimmuni e la malattia celiaca.
- Metabolismo lipidico e glucidico. Spesso si evidenziano elevati livelli di colesterolo ed aumentato livello di trigliceridi che sembra legato soprattutto all’obesità e all’iperinsulinismo. Un’alterata tolleranza glucidica è presente nel 10-34% dei soggetti con Sindrome di Turner. In epoca adulta più della metà delle donne ha insulino-resistenza e propensione a sviluppare diabete mellito tipo 2.
- Fegato. E’ molto frequente un incremento dei livelli degli enzimi epatici con un aumentato rischio di steatosi epatica. Studi recenti, con biopsie epatiche eseguite in soggetti con elevazione persistente dei tests epatici, hanno evidenziato anomalie multiple come fibrosi, infiltrati infiammatori e in alcuni casi iperplasia nodulare rigenerativa e cirrosi.
- Accrescimento. La bassa statura è presente nel 95% dei casi. In queste pazienti si osserva un decremento della crescita staturale durante l’infanzia e non si verifica lo spurt accrescitivo puberale, per la mancanza di ormoni sessuali prodotti dall’ovaio. In età aduta, la statura media è di circa 142.5 cm (risultati del Gruppo Italiano di Studio sulla Sindrome di Turner), circa 20 cm inferiore rispetto alla popolazione femminile di riferimento. La causa del ritardo di crescita non è nota, ma si pensa che si tratti di un difetto primitivo dell’osso. Geni determinanti la statura sono localizzati sul braccio corto del cromosoma X (Xp22) e Y (Yp11).
- Gonadi. I soggetti con sindrome di Turner presentano frequentemente disgenesia gonadica e circa il 90 % dei soggetti va incontro ad insufficienza ovarica, pertanto solo il 5-10% ha una funzionalità ovarica sufficiente ad innescare una normale pubertà: in pochi casi si hanno mestruazioni spontanee, che peraltro persistono per un periodo di tempo limitato.
Leggi anche:
- Le malattie genetiche più diffuse al mondo
- Sindrome di Klinefelter: cariotipo, cause, sintomi e cura
- Sindrome di Down: cause, sintomi in gravidanza e nei neonati
Prognosi
L’aspettativa di vita dei soggetti, che giungono alla nascita, sembra essere sovrapponibile a quella della popolazione generale. Buona parte degli embrioni con cariotipo 45,X (presente nell’1,5% dei concepimenti) va incontro ad aborto spontaneo; in questi casi è frequente la presenza di igroma cistico allecografia prenatale.
Test diagnostici
La diagnosi è facilmente effettuabile mediante l’esecuzione di una analisi del cariotipo, che mette in evidenza l’assenza di uno dei due cromosomi X; nei casi di monosomia completa, tutte le cellule presentano tale assetto, mentre nei soggetti con mosaicismosolo una quota variabile di cellule è coinvolta. Nei rimanenti casi è possibile mettere in evidenza la presenza di modificazioni della struttura, definiti riarrangiamenti, del cromosoma X.
Diagnosi differenziale
Questa condizione entra in diagnosi differenziale con la sindrome di Noonan, dove sono presenti sia la bassa statura sia alcune caratteristiche fenotipiche sovrapponibili, quali l’impianto dei capelli e lo pterigio del collo. In tale condizione non sono comunque presenti anomalie cromosomiche del cromosoma X ed anche i maschi possono esserne affetti.
Terapia
Nei soggetti con deficit accrescitivo, l’utilizzo dell’ormone della crescita biosintetico, ad alte dosi e per lunghi periodi di tempo, determina un buon guadagno staturale e un miglioramento della statura definitiva. Molte delle ragazze trattate con GH superano i 150 cm potendosi porre nel range di normalità. Anche se non è stata stabilita l’età ottimale per l’inizio della terapia, dati preliminari indicano che un inizio molto precoce è più efficace, non rischioso e permette di poter progredire nella pubertà ad una età fisiologica. La terapia con GH biosintetico, peraltro prevista dal SSN, va proseguita fino al raggiungimento della statura definitiva e necessita di regolari controlli clinici, laboratoristici ed endocrinologici. All’età della pubertà se non compaiono segni spontanei e’ consigliato l’utilizzo di terapia sostitutiva estrogenica, per garantire lo sviluppo dei caratteri sessuali secondari e per mantenere una buona integrità tessutale e ossea. L’induzione della pubertà con estrogeni deve riflettere il normale processo puberale e lo schema terapia deve essere individualizzato. Studi recenti hanno evidenziato che l’inizio degli estrogeni a circa 12 anni non interferisce con l’effetto positivo del GH sulla crescita, se il GH è stato somministrato per un periodo sufficientemente lungo. Il trattamento con terapia sostitutiva estrogenica si è dimostrato inoltre efficace nel ridurre la pressione diastolica, nel migliorare l’elasticità arteriosa e la funzionalità epatica.
Leggi anche:
- Sindrome di Edwards (trisomia 18): cause, sintomi, diagnosi, cure
- Sindrome di Patau (trisomia 13): cause, sintomi, diagnosi, cure
- Fibrosi cistica polmonare: cos’è, sintomi in neonati e bambini, cure
- Malattia di Huntington: cos’è, ereditarietà, come si trasmette, età di insorgenza
- Anemia falciforme: cosa significa, cause, sintomi e cure
- Differenze tra la distrofia muscolare di Duchenne e di Becker
- Talassemia: cos’è, sintomi, cure, differenti tipi ed alimentazione
- Celiachia: cos’è il glutine, in quali alimenti è contenuto ed in quali no?
- Sindrome di Noonan: cause, sintomi nel neonato, aspettative di vita
- Sindrome di Bloom: cause, sintomi, diagnosi e terapia
- Consulenza genetica, diagnosi prenatale, amniocentesi, esame dei villi coriali, tri test, screening GUIDA COMPLETA
- Test genetici diagnostici, screening neonatale esteso e test genetici prenatali
- Ecografie 3D e 4D: a cosa servono e quali sono le differenze con l’ecografia standard?
- Duo test in gravidanza: settimana, risultati, rischi, procedura, costo
- Tri test in gravidanza: settimana, risultati, rischi, procedura, costo
- Quad test in gravidanza: settimana, risultati, rischi, procedura, costo
- Translucenza nucale in gravidanza: a cosa serve, quando si fa, a chi è consigliata?
- Test combinato (duo test più translucenza nucale) in gravidanza
- Test integrato in gravidanza: a cosa serve, quando si fa, a chi è consigliato?
- Test integrato sierico: a cosa serve, quando si fa, a chi è consigliato?
- Differenze tra duo test, tri test, quad test, translucenza nucale, amniocentesi, villocentesi, test combinato e integrato
- Analisi del cariotipo, cariotipo normale e patologico, amniocentesi, villocentesi
- Amniocentesi precoce e tardiva: settimana, risultati, rischi, procedura, dolore, costo
- Analisi dei villi coriali (villocentesi): settimana, risultati, rischi, procedura, dolore, costo
- Differenze tra amniocentesi e villocentesi (prelievo dei villi coriali) vantaggi, svantaggi
- Cos’è un cromosoma ed a che serve?
- Differenza tra allele dominante e recessivo
- Differenza tra omozigote ed eterozigote
- Differenza tra gene e allele
- Sindrome dell’idiota sapiente: cause, caratteristiche e sintomi
- Sindrome del tramonto o del crepuscolo: cause, sintomi e cura
- Ritardo mentale nei bambini lieve, moderato, grave: si guarisce?
- Che cos’è l’intelligenza umana: definizione, significato e psicologia
- Quoziente d’intelligenza: valori, significato, test ed ereditarietà
- Problem solving: cos’è, caratteristiche, tecniche, fasi ed esempi
- Sindrome di Tourette: cause, sintomi, diagnosi e trattamento
- Sindrome di Tourette: si può guarire definitivamente? Come si guarisce?
- Differenza tra genotipo e fenotipo
- Quanti cromosomi hanno esseri umani, scimmie, cani, gatti e topi?
- Quanti cromosomi ha chi è affetto da Sindrome di Down?
- Cos’è un gene ed a che serve?
- Cosa sono gli alleli ed a che servono?
- Differenza tra cellule eucariote e procariote
- Virus e virioni: cosa sono, come sono fatti, come funzionano e come si riproducono
- Differenza tra cellula aploide e diploide con esempi
- Riproduzione cellulare e ciclo cellulare
- Meiosi: spiegazione di tutte tappe
- Mitosi: spiegazione delle quattro fasi
- Differenza tra mitocondri e cloroplasti
Lo Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o unisciti al nostro gruppo Facebook o ancora seguici su Twitter, su Instagram o su Pinterest, grazie!
