
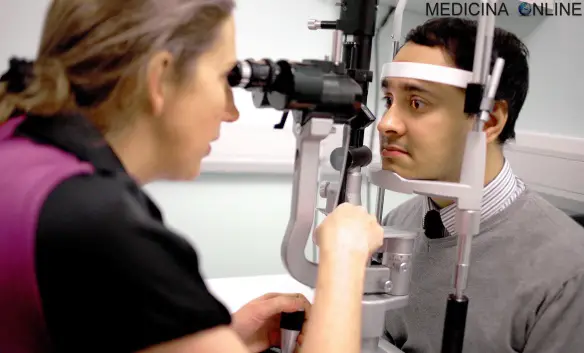
La sindrome di Behçet o “BD” (in inglese “Behçet’s disease”) è una rara sindrome di origine sconosciuta, probabilmente autoimmunitaria, caratterizzata da un quadro infiammatorio multisistemico recidivante con afte orali, genitali, uveite e tromboflebite, con frequente coinvolgimento delle articolazioni, della cute, del sistema nervoso centrale e del tratto gastrointestinale. Qualora venga coinvolto il sistema nervoso, si parla di “neuro-BD”. La sindrome di Behçet interessa varie figure professionali, tra cui dermatologo, odontoiatra ed immunologo.
Denominazione
La sindrome di Behçet deve il suo nome al medico dermatologo turco Hulusi Behçet, che per primo la descrisse nel 1937.
Epidemiologia
La prevalenza di questa sindrome è 1-9 ogni 100000. In Italia l’incidenza è di 2,4 casi ogni milione di abitanti e la prevalenza di 3,8/100000. La prevalenza più elevata è segnalata in Turchia essendo superiore ad 1 su 1000. L’età di esordio è variabile e può interessare età adulta, adolescenti ed infanzia, tuttavia colpisce prevalentemente tra i 25 ed i 30 anni.
Cause e fattori di rischio
La sindrome di Behçet ha eziologia attualmente sconosciuta; alcuni studi ipotizzano un’associazione genetica con l’antigene HLA-B51 (e con l’HLA-B57 limitatamente alla popolazione caucasica): tale antigene si associa infatti alla sindrome nel 50-70% dei pazienti. Sembra inoltre che i livelli anomali delle citochine (come IL-6, TNF-a, IL-8, IL-12, IL-17 e IL-21) abbiano un ruolo nella sua patogenesi, che quindi sarebbe autoimmunitaria. Si suppone inoltre che alcune infezioni (come ad esempio quelle provocate dal batterio Streptococcus sanguis) in associazione ad altre cause ambientali non ancora ben comprese, possano innescarsi sulla predisposizione genetica, scatenando i sintomi della sindrome, che sarebbe quindi ad eziologia “multifattoriale”.
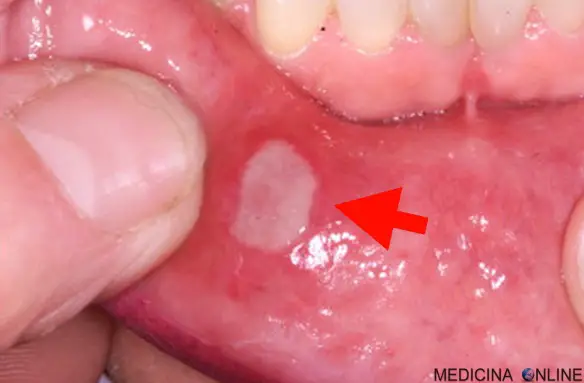
Afta orale
Sintomi e segni
L’esordio avviene di solito intorno ai 30 anni, ma sono stati descritti anche casi pediatrici. Uno dei segni della sindrome sono le afte orali recidivanti, tonde, con bordi sopraelevati eritematosi ben distinti e con diametro da uno a 3 cm. Alle afte orali si possono spesso associare le afte genitali. Altri possibili sintomi e segni, sono:
- pseudo-follicolite;
- eritema nodoso;
- uveite posteriore;
- vasculite retinica;
- riduzione dell’acuità visiva e perdita permanente della vista nel 20% dei casi;
- dolore oculare;
- arrossamento oculare;
- ipopion;
- corpi galleggianti nel campo visivo;
- artralgia;
- artrite;
- febbre alta intermittente all’esordio della malattia e febbricola serale nella fase cronica;
- stanchezza marcatissima fin dal mattino (astenia);
- vasculite del sistema venoso, con trombosi nel distretto femorale-iliaco, nella vena cava inferiore e cava superiore e nei territori cerebrali;
- trombosi arteriose e aneurismi soprattutto nei vasi polmonari;
- segni neurologici sporadici (cefalea, segni piramidali con emiparesi, alterazioni del comportamento e disfunzioni sfinteriche);
- lesioni ulcerative e/o aftoidi possono nel tratto digestivo, soprattutto nella regione ileo-cecale e nel colon ascendente, con eventuali emorragie e perforazioni che possono portare a sanguinamento occulto nelle feci ed anemia ferrocarenziale.
- ulcerazione genitale ricorrente
- lesioni oculari
- patergia (iperreattività cutanea a stimoli comuni con comparsa di papule e pustole).
Leggi anche:
Diagnosi
La sindrome di Behçet non è diagnosticabile né da sintomi patognomici né da esami di laboratorio. Importanti sono l’anamnesi (raccolta dei dati e dei sintomi del paziente) e l’esame obiettivo (visita vera e propria con raccolta dei segni). Come accennato già nel paragrafo precedente, per effettuare la diagnosi si procede seguendo dei criteri diagnostici: i pazienti devono presentare (in assenza di altre patologie che giustifichino questi segni e sintomi) un’ulcerazione orale ricorrente (aftosa o erpetiforme) almeno 3 volte in un periodo di 12 mesi con l’aggiunta di almeno due o più di questi criteri:
- ulcerazione genitale ricorrente;
- lesioni oculari:
- uveite anteriore;
- uveite posteriore;
- vasculite retinica;
- lesioni cutanee:
- eritema nodoso;
- follicolite;
- lesioni papulo-pustolose o noduli acneiformi in pazienti in età post-adolescenziale che non assumano corticosteroidi.
La positività dell’antigene HLA-B51 ha valore solo qualora serva per confermare la diagnosi in caso si manifestino soltanto due dei tre sintomi necessari.
Diagnosi nella neuro-BD
Qualora siano presenti anche sintomi nervosi (neuro-BD) è necessario anche effettuare la puntura lombare. La risonanza magnetica può inoltre rivelare lesioni infiammatorie nel tronco encefalico e nelle aree emisferiche.
Diagnosi differenziale
- amiloidosi;
- sindrome da anticorpi antifosfolipidi;
- malattie infiammatorie croniche intestinali come la malattia di Crohn;
- sindromi paraneoplastiche;
- poliarterite nodosa;
- lupus eritematoso sistemico;
- uveite infettiva;
- febbre periodica con stomatite aftosa, faringite e adenite” (PFAPA);
- policondrite ricorrente;
- sarcoidosi;
- stomatite aftosa recidivante;
- arterite di Takayasu;
- sclerosi multipla.
Terapie
- alleviare i sintomi;
- ridurre l’infiammazione;
- modulare il sistema immunitario.
La terapia con inibitori del TNF-alfa, come l’infliximab, mostra buoni risultati nel trattamento dell’uveite associata con la malattia. Un altro farmaco anti TNF-alfa, l’etanercept, è utile nei pazienti con sintomi prevalentemente cutanei e delle mucose. L’interferone alfa-2a rappresentare un efficace trattamento alternativo, specie per le ulcere genitali e per le lesioni oculari. L’azatioprina usata in combinazione con interferone alfa-2b presenta buoni risultati; la colchicina è utile per il trattamento di ulcere genitali, eritema nodoso e artrite. La somministrazione di immunoglobuline endovena potrebbe essere un trattamento valido per i casi più gravi. La somministrazione di alte dosi di corticosteroidi (1 mg/kg/die di prednisone orale) è indicata in caso di gravi manifestazioni della malattia.
Leggi anche:
- Stomatite aftosa recidivante: cause, sintomi, cure, collutorio, cosa mangiare
- Cheilite angolare (boccarola): cause, sintomi, diagnosi, cura, rimedi
- Febbre periodica con stomatite aftosa, faringite e adenite (PFAPA)
Prognosi
La prognosi per la sindrome di Behçet è correlata a vari fattori come età del paziente, eventuale presenza di altre patologie (ad esempio diabete o ipertensione arteriosa), sito di manifestazione, gravità e rapidità nella diagnosi: in particolare la diagnosi precoce di questa sindrome è fondamentale per avviare rapidamente la terapia idonea, che conduce spesso alla risoluzione della sintomatologia; al contrario in caso di ritardo nella diagnosi o assenza di trattamento, la prognosi è grave, specie a causa di:
- coinvolgimento oculare che può portare alla cecità completa;
- rischio di ictus o della rottura di un’arteria, che può portare ad emorragia rapidamente mortale;
- sintomi neurologici che possono esitare in un’encefalopatia grave, deficit motori e/o sensitivi e perdita parziale o totale dell’autonomia.
Una terapia oftalmica intensiva associata a trattamenti immunosoppressivi sembra comunque ridurre molto la morbilità. Circa il 5% dei pazienti, muore a distanza di 8 anni dalla diagnosi; la mortalità a 1 anno dalla diagnosi è dell’1,2% mentre quella a 5 anni è del 3,3%. L’età media dei decessi è di circa 34 anni. Il 40% dei decessi è causato da patologie dei vasi sanguigni, in particolare da rottura di aneurisma delle arterie polmonari. I maschi hanno mediamente una prognosi peggiore rispetto alle donne.
Leggi anche:
- Afte, nevralgia, herpes ed altre cause di dolore alla lingua (glossodinia)
- Uveiti: classificazione, cause, sintomi, diagnosi e terapie
- Tromboflebite superficiale: significato, diagnosi, cure, rischi
- Lingua bianca e afte nei neonati: cause e cure
- Papille gustative della lingua: tipi, gusti, distribuzione e funzioni
- Papille gustative ingrossate: quando la lingua è infiammata
- Igiene orale: consigli e prodotti per pulire lingua, denti e bocca
- Collutori naturali e fatti in casa per disinfettare la lingua
- Cibi che macchiano i denti: quali evitare ed i consigli per mantenerli bianchi
- Dolore alla lingua ed alla bocca: cause, complicanze, dieta e terapia
- Alitosi: il tipo di odore del tuo alito cattivo rivela la patologia che hai
- Alito cattivo: tutti i rimedi migliori per combattere l’alitosi
- Come pulire efficacemente la lingua in modo semplice e sicuro
- Come e quando iniziare a lavare i denti ai bambini?
- Quante volte e quando lavare i denti ogni giorno?
- Lavare i denti correttamente: per quanto tempo?
- Quale dentifricio usare per lavarsi i denti?
- Come utilizzare correttamente il filo interdentale?
- Come lavare i denti ai bambini in modo facile e corretto
- Come lavarsi i denti in modo corretto [VIDEO]
- Come lavare i denti senza dentifricio
- Come lavarsi i denti senza spazzolino
- Come lavarsi i denti con l’apparecchio
- Come pulire l’apparecchio per i denti mobile e fisso
- Lingua infiammata (glossite): cause, sintomi, terapie, rimedi
- Lingua a carta geografica in bambini e adulti: stress, rimedi, quanto dura
- Glossite atrofica: cause, diagnosi, consigli e terapie
- Lingua bianca, impastata, spaccata: cause e quando è pericolosa
- In quali casi la lingua viene amputata?
- Lingua gialla con alito cattivo o mal gola: cause e rimedi
- Patina bianca sulla lingua: da cosa può essere causata?
- La lingua dice molto sulla tua salute: ecco come “leggerla”
- Lingua verde: cause e terapie
- Lingua nera villosa: cause, diagnosi e terapie
- La lingua che parli influenza la tua personalità ed il tuo cervello
- Cos’è la lingua: anatomia e funzioni in sintesi
- Differenze tra allergia alimentare ed intolleranza alimentare
- Bocca secca ed asciutta da ansia, diabete, malattie del fegato: diagnosi e cure
- Salivazione eccessiva dopo i pasti, in gravidanza, nei bambini: quali rimedi?
- Ghiandole salivari ingrossate: sintomi, cause, come si curano
- Ghiandole salivari: anatomia e funzioni in sintesi
- Tumore delle ghiandole salivari: sintomi, diagnosi e terapie
- Dolore alla mandibola: cause e sintomi
- Articolazione temporo mandibolare (ATM): anatomia e funzioni
- Sindrome temporo mandibolare: sintomi, diagnosi e cure
- Differenza tra mascella e mandibola: sono sinonimi?
- Di cosa è fatta la saliva, quanta ne produciamo, a che serve?
- Labbra blu: da cosa sono causate e come si curano in bimbi ed adulti
- Labbra screpolate e gonfie: cause e rimedi in bambini ed adulti
- Herpes labiale: cause, sintomi, rimedi e trattamento farmacologico
- Labbra gonfie (gonfiore labiale): possibili cause, sintomi e rimedi
- Cattivo sapore in bocca acido o amaro: rimedi e quando è pericoloso
- Denti sensibili: sbiancamento, caldo, freddo, collutorio ed altre cause
- La lingua è un muscolo?
- Di cosa è fatta la lingua?
- Lingua: anatomia e funzioni in sintesi
- Frenulo linguale: cos’è e cosa fare se è troppo corto
- Quali sono i muscoli che compongono la lingua?
- Glossodinia, sindrome della bocca che brucia: sintomi e cure
- Gengive: anatomia, funzioni e patologie più diffuse in sintesi
- Gengivite: cause, patogenesi, sintomi e terapie
- Denti: anatomia, funzioni e patologie più diffuse in sintesi
- Guance: anatomia e funzioni in sintesi
- Labbra: anatomia, funzioni, patologie e significato sociale
- Palato: anatomia, funzioni e patologie più diffuse in sintesi
- Palatoschisi: cause, problemi derivanti e cure
- Labbro leporino (cheiloschisi): tipi, cause, problematiche e cure
- Differenza tra palatoschisi e labbro leporino
- A quale sapore corrisponde il gusto umami?
- Glutammato monosodico: cos’è ed in quali cibi trovarlo
- Glutammato e sindrome del ristorante cinese: effetti sulla salute
Dott. Emilio Alessio Loiacono
Medico Chirurgo
Direttore dello Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o unisciti al nostro gruppo Facebook o ancora seguici su Twitter, su Instagram, su Mastodon, su Tumblr e su Pinterest, grazie!
 L’encefalite da anticorpi anti-NMDA è una rara forma di encefalite (infiammazione dell’encefalo), a patogenesi autoimmunitaria solo di recente descritta (nel 2007), che colpisce prevalentemente giovani donne di età inferiore ai 30 anni, più raramente uomini, bambini ed anziani. La patologia è
L’encefalite da anticorpi anti-NMDA è una rara forma di encefalite (infiammazione dell’encefalo), a patogenesi autoimmunitaria solo di recente descritta (nel 2007), che colpisce prevalentemente giovani donne di età inferiore ai 30 anni, più raramente uomini, bambini ed anziani. La patologia è  Con “colangite sclerosante primitiva” (o colangite sclerosante idiopatica) in campo medico, si intende un complesso di disturbi che riguarda i dotti biliari, con
Con “colangite sclerosante primitiva” (o colangite sclerosante idiopatica) in campo medico, si intende un complesso di disturbi che riguarda i dotti biliari, con  La sindrome di Sjögren (in inglese “Sjögren’s syndrome”) è una malattia infiammatoria cronica su base autoimmune, caratterizzata dalla distruzione di ghiandole esocrine (ghiandole salivari minori, ghiandole lacrimali, parotidi) mediata dai linfociti T. C’è inoltre un’eccessiva attivazione dei linfociti B con produzione di autoanticorpi quali fattore reumatoide (FR), anti SS-A/Ro e anti SS-B/La. La malattia è caratterizzata da periodi di stasi alternati ad altri di riacutizzazione. Può essere anche il primum movens a distanza di anni di una malattia immunitaria più grave, spesso dell’artrite reumatoide.
La sindrome di Sjögren (in inglese “Sjögren’s syndrome”) è una malattia infiammatoria cronica su base autoimmune, caratterizzata dalla distruzione di ghiandole esocrine (ghiandole salivari minori, ghiandole lacrimali, parotidi) mediata dai linfociti T. C’è inoltre un’eccessiva attivazione dei linfociti B con produzione di autoanticorpi quali fattore reumatoide (FR), anti SS-A/Ro e anti SS-B/La. La malattia è caratterizzata da periodi di stasi alternati ad altri di riacutizzazione. Può essere anche il primum movens a distanza di anni di una malattia immunitaria più grave, spesso dell’artrite reumatoide.