L’aceruloplasminemia è una rarissima patologia neurodegenerativa genetica contraddistinta da accumulo di ferro nel cervello causato dalla completa assenza dell’attività della ceruloplasmina ferrossidasi dovuta a mutazioni omozigoti del gene della ceruloplasmina. E’ caratterizzata da anemia associata a degenerazione della retina, diabete mellito e vari sintomi neurologici.
L’aceruloplasminemia è una rarissima patologia neurodegenerativa genetica contraddistinta da accumulo di ferro nel cervello causato dalla completa assenza dell’attività della ceruloplasmina ferrossidasi dovuta a mutazioni omozigoti del gene della ceruloplasmina. E’ caratterizzata da anemia associata a degenerazione della retina, diabete mellito e vari sintomi neurologici.
Epidemiologia
Colpisce una persona su un milione, esordisce in età adulta. Essendo una malattia estremamente rara, in letteratura scientifica sono stati descritti meno di 60 casi.
Cause
La aceruloplasminemia è una malattia genetica causata da mutazioni nel gene della ceruloplasmina, una proteina coinvolta nel metabolismo del ferro. Viene ereditata dai genitori e si trasmette con modalità autosomica recessiva.
Trasmissione autosomica recessiva
Una malattia è detta a trasmissione autosomica recessiva quando l’allele alterato deve essere presente in coppia (omozigosi), cioè sono necessarie due copie dell’allele difettoso per far sì che la malattia si esprima, a prescindere dal sesso. Non basta un solo genitore portatore sano o malato, bensì entrambi i genitori devono essere portatori sani o malati. Il fenotipo quindi si esprime quando nel genotipo dell’individuo sono presenti entrambi gli alleli responsabili, fatto che spiega l’alta probabilità di sviluppare malattie genetiche in caso di incesto. Quindi:
- un individuo che possegga entrambi gli alleli alterati: è portatore ed è malato;
- un individuo che possegga solo un allele alterato: è portatore ma è sano;
- un individuo che non possegga nessun allele alterato: NON è portatore ed è sano.
Essere portatore sano vuol dire quindi NON avere la patologia ma possedere nel proprio genotipo un allele mutato, che può essere trasmesso alle generazioni successive.
Dalla combinazione delle possibili condizioni di genitori sani, malati e portatori sani, deriva la distribuzione probabilità che la malattia sia trasmessa ai figli:
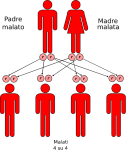
- genitori malato-malato: la probabilità che il figlio/a nasca malato è del 100%;
- genitori sano-malato: la probabilità che il figlio/a nasca portatore sano è del 100%;
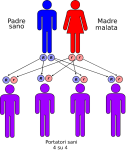
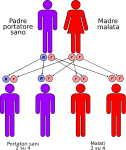
- genitori malato-portatore sano: la probabilità che il figlio/a nasca malato è del 50% e del 50% che nasca portatore sano;
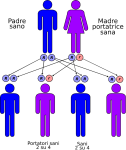
- genitori sano-portatore sano: la probabilità che il figlio/a nasca sano è del 50% e del 50% che nasca portatore sano;
- genitori portatore-portatore: la probabilità che il figlio/a nasca portatore sano è del 50% mentre è del 25% che nasca sano o malato.
Se nessuno dei genitori ha un allele mutato, non c’è ovviamente alcuna trasmissione autosomica recessiva ed i figli saranno tutti sani e NON portatori dell’allele mutato.
Nell’immagine che segue, è raffigurata la tipica situazione in cui entrambi i genitori sono sani ma portatori dell’allele mutato:
- un figlio su quattro avrà entrambi gli alleli alterati e sarà malato ed ovviamente portatore;
- due figli su quattro avranno un allele normale ed uno alterato e saranno sani ma anche portatori;
- un figlio su quattro avrà entrambi gli alleli normali e sarà sano e NON portatore.

Le altre quattro situazioni possibili sono raffigurate nelle seguenti immagini:
Sintomi e segni
Come già in parte prima accennato, l’aceruloplasminemia si manifesta nell’adulto con degenerazione retinica, diabete mellito e anemia. L’anemia nella aceruloplasminemia è refrattaria al trattamento con il ferro. Altri sintomi sono di tipo neurologico:
- atassia cioè mancanza di coordinazione muscolare che rende difficoltoso eseguire i movimenti volontari (mancanza di coordinazione fra tronco e braccia, tronco e capo, incoordinazione dei movimenti dell’occhio, incontinenza, difficoltà di deglutizione);
- movimenti involontari (blefarospasmo, contrazioni del viso, distonia della faccia e del collo, tremori e corea);
- parkinsonismo (tremore tremore a riposo che diminuisce con i movimenti volontari e scompare durante il sonno);
- depressione;
- deficit cognitivo.
Diagnosi
La diagnosi di laboratorio si basa su:
- assenza della ceruloplasmina sierica;
- bassa concentrazione sierica di rame e di ferro;
- alta concentrazione sierica di ferritina.
La diagnosi è suggerita inoltre dal riscontro alla risonanza magnetica cerebrale di una caratteristica anomala bassa intensità di segnale, che riflette l’accumulo di ferro nel cervello (striato, talamo, nucleo dentato) e nel fegato, nelle immagini T1, T2 pesate. L’analisi genetica può confermare la diagnosi. La diagnosi differenziale si pone con le altre forme di NBIA a esordio tardivo, lentamente progressive, compresa la neurodegenerazione atipica associata al difetto di pantotenato-chinasi (PKAN), la neuroferritinopatia, l’emocromatosi ereditaria, la malattia di Wilson, la malattia di Huntington, l’atrofia dentato-rubro-pallido-luisiana (DRPLA), la malattia di Parkinson giovanile, le atassie spinocerebellari ereditarie (si vedano questi termini) e gli effetti o la tossicità da farmaci.
Diagnosi prenatale
La diagnosi prenatale per le gravidanze ad aumentato rischio può essere disponibile presso laboratori specializzati, nei casi in cui la mutazione responsabile della malattia sia stata preventivamente identificata in un soggetto affetto della famiglia.
Trattamento
Il trattamento si basa sulla somministrazione orale o endovenosa di chelanti del ferro (deferiprone o deferasirox), che producono un miglioramento del diabete e dei sintomi neurologici. La combinazione di desferoxamina e plasma umano fresco congelato (FFP) contribuisce a ridurre il contenuto epatico di ferro. Gli antiossidanti, come la vitamina E, e la somministrazione orale di zinco possono prevenire il danno tissutale.
Prognosi
La prognosi è legata all’insufficienza cardiaca da accumulo di ferro nell’organo. Sono noti 5 pazienti con aceruloplasminemia deceduti per insufficienza cardiaca, probabilmente secondaria all’accumulo intracardiaco di ferro in un arco di 60 anni. In assenza di insufficienza cardiaca e con un adeguato trattamento del diabete, la prognosi è buona.
Leggi anche:
- Sovraccarico di ferro: emosiderosi ed emocromatosi
- Emocromatosi ereditaria e secondaria: sintomi, dieta, diagnosi, cure
- Anemia da carenza di ferro: cause, sintomi e cure
- Ferro: alimenti ricchi di ferro, fabbisogno giornaliero, integratore
- Sali minerali: definizione, funzioni, alimenti, integratori [GUIDA COMPLETA]
- Carenza, sovraccarico e tossicità da ferro: anemia ed emocromatosi
- Iodio: fabbisogno giornaliero, funzioni, dieta, carenza, tossicità
- Fluoro: carenza, tossicità, fabbisogno, dieta e funzioni
- Zinco: carenza, tossicità, fabbisogno, dieta, integratore e funzioni
- Cromo: benefici, funzioni, fabbisogno, dieta, integratore, carenza, tossicità
- Iodio: benefici, funzioni, fabbisogno, dieta, integratore, carenza, tossicità
- Selenio: benefici, funzioni, fabbisogno, dieta, integratore, carenza, tossicità
- Manganese: benefici, funzioni, fabbisogno, dieta, integratore, carenza, tossicità
- Molibdeno: benefici, funzioni, fabbisogno, dieta, integratore, carenza, tossicità
- Rame: benefici, funzioni, fabbisogno, dieta, integratore, carenza, tossicità
- Malattia di Wilson: dieta, sintomi, mortalità, aspettativa di vita
- Ipovitaminosi (carenza di vitamine): sintomi, lingua, unghie, cura
- Ipervitaminosi (eccesso di vitamine): sintomi ed esempi
- Malnutrizione per difetto o eccesso: definizione, sintomi, significato
- Ecco come il nostro corpo ci segnala la carenza di vitamine
- Hai voglia di un cibo in particolare? E’ il tuo corpo che ti rivela le carenze nutrizionali che hai
- Classifiche di calorie e di tutti i valori nutrizionali dei cibi
- Celiachia: cos’è il glutine, in quali alimenti è contenuto ed in quali no?
- Morbo di Crohn: cos’è, cause scatenanti, sintomi, cure e dieta
- Sindrome di Korsakoff e alcol: cause, sintomi, amnesia e cura
- Perdita di appetito: cause, ansia, stanchezza, tumore, cure
- Malassorbimento intestinale: esami, carboidrati, vitamine, dimagrimento
- Pellagra: significato, cause, subclinica, rischi, terapia, dieta consigliata
- Scorbuto: significato, pelle, sintomi, diagnosi, cura, immagini
- Carenza di vitamina A (retinolo): sintomi, dieta e integratori consigliati
- Carenza di vitamina D, rachitismo, osteomalacia: cause, sintomi, terapie, dieta, integratori
- Carenza di vitamina K: sintomi, fabbisogno, dieta e integratori consigliati
- Carenza di niacina e triptofano: pellagra, cause, terapie e dieta
- Carenza e dipendenza da vitamina B6: cause, sintomi, terapie, dieta, integratori
- Carenza di biotina: cause, sintomi, fabbisogno, dieta, integratori
- Carenza di vitamina C e scorbuto: cause, sintomi, cura, dieta, integratori
- Carenza di acido pantotenico (vitamina B5): cause, sintomi, cure, dieta, integratore
- Vitamina A (retinolo): a cosa serve, cosa provoca la sua carenza e quali alimenti ne sono ricchi?
- Una vostra amica è troppo magra? Vi insegno a capire se soffre di anoressia
- Alcolismo: test, cause, sintomi, definizione, effetti, danni, quando preoccuparsi
- Cachessia: oncologica, terminale, senile, significato e rimedi
- Gastrectomia parziale e totale: rischi, sopravvivenza, dieta
- Assumi abbastanza vitamina D? I sintomi che indicano la sua carenza e i cibi che ne contengono in abbondanza
- Beta carotene: cos’è, a cosa serve e in quali cibi lo trovo?
- Quali sono i cibi con più vitamina C, vitamina E, magnesio e beta carotene?
- Acido folico (vitamina B9): a cosa serve, in quali alimenti trovarlo e perché è importante prima e durante la gravidanza
- A che serve la vitamina B12? L’importanza in gravidanza e allattamento
- Perché cadono i capelli? Quanti capelli al giorno è normale perdere? E’ vero che i calvi hanno più testosterone?
- I tuoi capelli rivelano la tua salute: ecco come leggere i segnali che ti inviano
- Antiossidanti: alimenti ed integratori migliori contro i radicali liberi
- Radicali liberi: cosa sono, significato, invecchiamento, combatterli
- Radicali liberi: come combatterli e rimanere giovani
- Tutto sul Coenzima Q10: dove si trova, a cosa serve e dosaggi
- Astaxantina: benefici, integratore, controindicazioni, dosaggio
- Carnosina: benefici, integratore, controindicazioni, cibi, dose
- Bromelina: integratore, compresse, benefici, controindicazioni
- Polifenoli: integratori, significato, dove si trovano, benefici
- Flavonoidi: integratori, benefici, alimenti, controindicazioni
- Isoflavoni: integratori, alimenti, dove si trovano, controindicazioni
- Resveratrolo: benefici, integratori, dosaggio, alimenti
- Catechine: integratori, alimenti, dove si trovano, effetti collaterali
- Quercetina: integratore, alimenti, cibi, benefici, effetti collaterali
- Ginkgo biloba: a cosa serve, indicazioni, dosaggio, effetti collaterali
- Melatonina 1 e 2mg per insonnia: quando assumerla e controindicazioni
Lo Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o unisciti al nostro gruppo Facebook o ancora seguici su Twitter, su Instagram o su Pinterest, grazie!
Condividi questo articolo:

 In un villaggio della Repubblica Dominicana chiamato Salinas, accade da anni qualcosa di veramente strano. Tante bambine all’età 12 anni si trasformano letteralmente in ragazzi.
In un villaggio della Repubblica Dominicana chiamato Salinas, accade da anni qualcosa di veramente strano. Tante bambine all’età 12 anni si trasformano letteralmente in ragazzi.