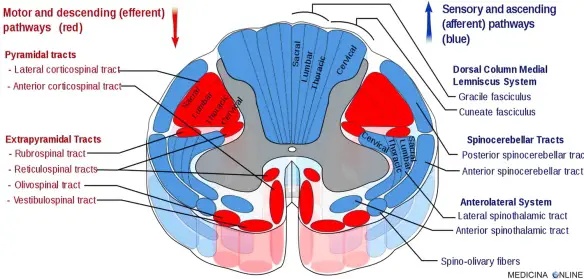
 Il movimento è un atto complesso che è il risultato dell’azione del sistema motorio piramidale e del sistema motorio extrapiramidale. In realtà Continua a leggere
Il movimento è un atto complesso che è il risultato dell’azione del sistema motorio piramidale e del sistema motorio extrapiramidale. In realtà Continua a leggere
Differenza tra via piramidale ed extrapiramidale
Rispondi

Il movimento è un atto complesso che è il risultato dell’azione del sistema motorio piramidale e del sistema motorio extrapiramidale. In realtà Continua a leggere
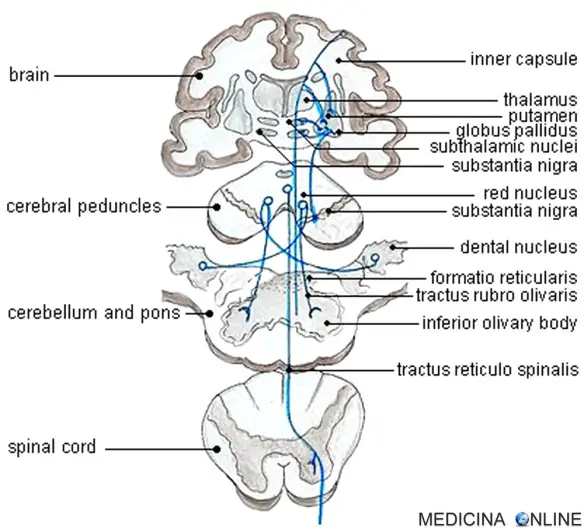 Il sistema extrapiramidale (o “via extrapiramidale“; in inglese “extrapyramidal system” o “extrapyramidal tracts“) è un insieme di vie e di centri nervosi che agiscono direttamente o Continua a leggere
Il sistema extrapiramidale (o “via extrapiramidale“; in inglese “extrapyramidal system” o “extrapyramidal tracts“) è un insieme di vie e di centri nervosi che agiscono direttamente o Continua a leggere
 La malattia (o morbo o còrea) di Huntington è una patologia ereditaria causata dalla degenerazione di neuroni situati in specifiche aree cerebrali – striato e corteccia cerebrale – e caratterizzata da una generale atrofia del cervello. I sintomi iniziali possono essere bruschi mutamenti dell’umore, apatia, irritabilità, depressione e rabbia, difficoltà nella guida, nell’imparare cose nuove o nel prendere una decisione. Altri possono presentare cambiamenti nella scrittura e movimenti involontari delle dita, dei piedi, del viso o del tronco (chiamati “còrea” dal termine greco che significa “danza”). In altri casi possono insorgere disturbi dell’equilibrio e del coordinamento motorio con accentuato rischio di cadute. L’ordine di comparsa di questi sintomi e la gravità possono variare notevolmente da un individuo all’altro, così come l’età d’insorgenza.
La malattia (o morbo o còrea) di Huntington è una patologia ereditaria causata dalla degenerazione di neuroni situati in specifiche aree cerebrali – striato e corteccia cerebrale – e caratterizzata da una generale atrofia del cervello. I sintomi iniziali possono essere bruschi mutamenti dell’umore, apatia, irritabilità, depressione e rabbia, difficoltà nella guida, nell’imparare cose nuove o nel prendere una decisione. Altri possono presentare cambiamenti nella scrittura e movimenti involontari delle dita, dei piedi, del viso o del tronco (chiamati “còrea” dal termine greco che significa “danza”). In altri casi possono insorgere disturbi dell’equilibrio e del coordinamento motorio con accentuato rischio di cadute. L’ordine di comparsa di questi sintomi e la gravità possono variare notevolmente da un individuo all’altro, così come l’età d’insorgenza.
Nella sua forma più classica la malattia insorge tra i 35 e i 45 anni, ma in alcuni casi può manifestarsi prima dei 20 anni con un rapido declino del rendimento scolastico/lavorativo, cambiamenti nella scrittura, rigidità, tremori, lentezza e rapidi spasmi muscolari. La forma giovanile progredisce molto più rapidamente di quella adulta. La forma tardiva, infine, si manifesta dopo i 55 anni e può essere più complessa da diagnosticare per la compresenza di altre patologie, ma anche perché i sintomi possono essere particolarmente lievi e perciò più difficili da individuare.
La frequenza della malattia è stimata in 5-10/100.000 individui, ma non sempre viene correttamente diagnosticata: esiste perciò certamente un gran numero di portatori inconsapevoli.
La malattia si trasmette con modalità autosomico dominante: un genitore affetto ha cioè una probabilità del 50% di trasmetterla a ciascuno dei suoi figli, a prescindere dal sesso. Quello che viene trasmesso non è la malattia, ma la mutazione di un gene, IT-15, localizzato sul cromosoma 4 e codificante per la proteina huntingtina, dalle funzioni non ancora del tutto note ma comunque importante per lo sviluppo embrionale e nel cervello adulto. Il gene IT-15 sano presenta al suo interno una specifica sequenza CAG che si ripete fino a un massimo di 35 volte, ma che in presenza della mutazione si ripete per un numero eccessivo di volte (da 39 a oltre 100). Ogni individuo che eredita il gene mutato svilupperà più o meno precocemente la malattia.
Dal 1993, con la scoperta del gene responsabile, è disponibile il test genetico per diagnosticare la malattia di Huntington. Su un prelievo di sangue di un individuo a rischio ma ancora del tutto privo di sintomi è possibile condurre un’analisi genetica che consente di accertare la presenza o l’assenza della mutazione nel gene IT-15. La decisione di sottoporsi al test è molto delicata, perché il risultato coinvolge non solo il singolo individuo, ma anche la sua famiglia (coniuge, figli, fratelli). È perciò consigliabile fare riferimento a centri di ricerca che dispongono di professionisti idonei. Data l’attuale impossibilità di prevenire la malattia non vengono effettuati test sui minori di 18 anni. Inoltre la diagnosi corretta si basa non solo sul test genetico, ma anche sulla storia familiare del paziente e sulle immagini del cervello che si possono ottenere con TC e risonanza magnetica nucleare: vista la variabilità della sintomatologia, anche in base alla fase e alla durata della malattia, è fondamentale considerare le informazioni fornite dai vari strumenti disponibili e non da un solo elemento (per esempio un paziente in fase iniziale può avere TC e risonanza magnetica normali).
Purtroppo non esiste al momento una terapia risolutiva. I principali sintomi motori e psichiatrici possono essere tenuti sotto controllo con farmaci in grado di migliorare la qualità di vita del malato e prevenire eventuali complicazioni, ma non di guarire la malattia o interromperne il decorso. Sono tuttavia in corso diverse sperimentazioni cliniche per valutare l’efficacia di varie sostanze.
Leggi anche:
Lo Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o unisciti al nostro gruppo Facebook o ancora seguici su Twitter, su Instagram o su Pinterest, grazie!
 L’aceruloplasminemia è una rarissima patologia neurodegenerativa genetica contraddistinta da accumulo di ferro nel cervello causato dalla completa assenza dell’attività della ceruloplasmina ferrossidasi dovuta a mutazioni omozigoti del gene della ceruloplasmina. E’ caratterizzata da anemia associata a degenerazione della retina, diabete mellito e vari sintomi neurologici.
L’aceruloplasminemia è una rarissima patologia neurodegenerativa genetica contraddistinta da accumulo di ferro nel cervello causato dalla completa assenza dell’attività della ceruloplasmina ferrossidasi dovuta a mutazioni omozigoti del gene della ceruloplasmina. E’ caratterizzata da anemia associata a degenerazione della retina, diabete mellito e vari sintomi neurologici.
Colpisce una persona su un milione, esordisce in età adulta. Essendo una malattia estremamente rara, in letteratura scientifica sono stati descritti meno di 60 casi.
La aceruloplasminemia è una malattia genetica causata da mutazioni nel gene della ceruloplasmina, una proteina coinvolta nel metabolismo del ferro. Viene ereditata dai genitori e si trasmette con modalità autosomica recessiva.
Una malattia è detta a trasmissione autosomica recessiva quando l’allele alterato deve essere presente in coppia (omozigosi), cioè sono necessarie due copie dell’allele difettoso per far sì che la malattia si esprima, a prescindere dal sesso. Non basta un solo genitore portatore sano o malato, bensì entrambi i genitori devono essere portatori sani o malati. Il fenotipo quindi si esprime quando nel genotipo dell’individuo sono presenti entrambi gli alleli responsabili, fatto che spiega l’alta probabilità di sviluppare malattie genetiche in caso di incesto. Quindi:
Essere portatore sano vuol dire quindi NON avere la patologia ma possedere nel proprio genotipo un allele mutato, che può essere trasmesso alle generazioni successive.
Dalla combinazione delle possibili condizioni di genitori sani, malati e portatori sani, deriva la distribuzione probabilità che la malattia sia trasmessa ai figli:
Se nessuno dei genitori ha un allele mutato, non c’è ovviamente alcuna trasmissione autosomica recessiva ed i figli saranno tutti sani e NON portatori dell’allele mutato.
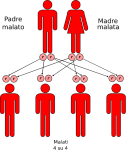
Nell’immagine che segue, è raffigurata la tipica situazione in cui entrambi i genitori sono sani ma portatori dell’allele mutato:

Le altre quattro situazioni possibili sono raffigurate nelle seguenti immagini:
Come già in parte prima accennato, l’aceruloplasminemia si manifesta nell’adulto con degenerazione retinica, diabete mellito e anemia. L’anemia nella aceruloplasminemia è refrattaria al trattamento con il ferro. Altri sintomi sono di tipo neurologico:
La diagnosi di laboratorio si basa su:
La diagnosi è suggerita inoltre dal riscontro alla risonanza magnetica cerebrale di una caratteristica anomala bassa intensità di segnale, che riflette l’accumulo di ferro nel cervello (striato, talamo, nucleo dentato) e nel fegato, nelle immagini T1, T2 pesate. L’analisi genetica può confermare la diagnosi. La diagnosi differenziale si pone con le altre forme di NBIA a esordio tardivo, lentamente progressive, compresa la neurodegenerazione atipica associata al difetto di pantotenato-chinasi (PKAN), la neuroferritinopatia, l’emocromatosi ereditaria, la malattia di Wilson, la malattia di Huntington, l’atrofia dentato-rubro-pallido-luisiana (DRPLA), la malattia di Parkinson giovanile, le atassie spinocerebellari ereditarie (si vedano questi termini) e gli effetti o la tossicità da farmaci.
La diagnosi prenatale per le gravidanze ad aumentato rischio può essere disponibile presso laboratori specializzati, nei casi in cui la mutazione responsabile della malattia sia stata preventivamente identificata in un soggetto affetto della famiglia.
Il trattamento si basa sulla somministrazione orale o endovenosa di chelanti del ferro (deferiprone o deferasirox), che producono un miglioramento del diabete e dei sintomi neurologici. La combinazione di desferoxamina e plasma umano fresco congelato (FFP) contribuisce a ridurre il contenuto epatico di ferro. Gli antiossidanti, come la vitamina E, e la somministrazione orale di zinco possono prevenire il danno tissutale.
La prognosi è legata all’insufficienza cardiaca da accumulo di ferro nell’organo. Sono noti 5 pazienti con aceruloplasminemia deceduti per insufficienza cardiaca, probabilmente secondaria all’accumulo intracardiaco di ferro in un arco di 60 anni. In assenza di insufficienza cardiaca e con un adeguato trattamento del diabete, la prognosi è buona.
Leggi anche:
Lo Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o unisciti al nostro gruppo Facebook o ancora seguici su Twitter, su Instagram o su Pinterest, grazie!
