 Il linfoma non Hodgkin è un tumore che nasce nel sistema linfatico e si sviluppa dai linfociti, cellule presenti nel sangue e nel tessuto linfatico di linfonodi, milza, timo, midollo osseo, tonsille e altre piccole aree dell’organismo. Invece di combattere le malattie, i linfociti (linfociti B o linfociti T) si accumulano nei linfonodi e in altri organi. Il linfoma non Hodgkin ha un’incidenza di 5 volte maggiore rispetto al linfoma di Hodgkin, e il 95% dei pazienti colpiti da questa malattia sono adulti. Sono state identificate almeno 30 forme diverse di questo tipo di tumore.
Il linfoma non Hodgkin è un tumore che nasce nel sistema linfatico e si sviluppa dai linfociti, cellule presenti nel sangue e nel tessuto linfatico di linfonodi, milza, timo, midollo osseo, tonsille e altre piccole aree dell’organismo. Invece di combattere le malattie, i linfociti (linfociti B o linfociti T) si accumulano nei linfonodi e in altri organi. Il linfoma non Hodgkin ha un’incidenza di 5 volte maggiore rispetto al linfoma di Hodgkin, e il 95% dei pazienti colpiti da questa malattia sono adulti. Sono state identificate almeno 30 forme diverse di questo tipo di tumore.
I linfomi possono nascere da tutti i tipi di linfociti (B, T e NK): i più comuni nel nostro Paese sono i linfomi di derivazione dai linfociti B. Le cause di questo tipo di tumore non sono ancora del tutto chiare. Si sa che alla base vi è un’anormale produzione di linfociti. In condizioni non patologiche, questi seguono un normale ciclo di vita: i più vecchi muoiono mentre l’organismo ne produce di nuovi per sostituirli. Nel linfoma non Hodgkin, invece, i vecchi linfociti non muoiono, ma continuano ad aumentare e ad accumularsi all’interno dei linfonodi o di altri tessuti, che si ingrossano o si alterano nella loro struttura.
Quali sono i fattori di rischio per il linfoma non Hodgkin?
La causa precisa di questo tipo di linfoma non è ancora conosciuta, ma alcuni fattori che possono aumentare il rischio di ammalarsi sono:
- condizioni di immunodepressione (ad esempio in seguito a un trapianto d’organo o a infezione da HIV).
- alcune infezioni virali, tra cui HIV, epatite C e virus di Epstein-Barr (responsabile della mononucleosi infettiva).
- agenti chimici presenti ad esempio negli insetticidi.
- età (soprattutto dopo i 60 anni).
Come si può prevenire il linfoma non hodgkin?
Trattandosi di una malattia rara, non è purtroppo noto alcun modo per prevenire l’insorgenza del linfoma non Hodgkin, se non evitando l’esposizione ai fattori di rischio comuni a tutti i tipi di cancro.
Per approfondire:
Diagnosi
Adulti e bambini con sospetta diagnosi di linfoma non Hodgkin vengono innanzitutto sottoposti ad un accurato esame obiettivo: l’ingrossamento non dolente dei linfonodi di collo, ascelle o inguine è spesso l’unico segno di linfoma non Hodgkin agli stadi iniziali. Altri sintomi possono includere febbre, sudorazione notturna, spossatezza, perdita di peso, dolore o gonfiore addominale, prurito persistente e dolore toracico, tosse o difficoltà respiratoria, a seconda della sede di insorgenza della malattia.
Durante la visita il medico analizza dimensioni e consistenza di tutti i linfonodi, alterati e normali. Vengono quindi eseguite le analisi del sangue e delle urine per escludere un’infezione o un’altra malattia che possano essere la causa dell’ingrossamento dei linfonodi.
Il primo passo per la diagnosi di linfoma non Hodgkin è la biopsia, di parte o di tutto il linfonodo. Il tessuto prelevato viene analizzato dall’anatomopatologo, che oltre a porre la diagnosi di linfoma ne stabilisce il sottogruppo di appartenenza: in generale un linfoma può crescere lentamente (linfoma indolente o “a basso grado di malignità”) o rapidamente (linfoma aggressivo o “ad alto grado di malignità”).
Esiste poi il piccolo sottogruppo dei linfomi “acuti” o molto aggressivi, assimilabili per la loro velocità di crescita alle leucemie acute. Il linfoma non Hodgkin è classificato in circa 30 tipi diversi, sulla base di numerosi fattori, tra cui la derivazione del tumore dai linfociti B o dai linfociti T, le dimensioni e le modificazioni genetiche dei linfociti, le modalità di aggregazione delle cellule tumorali e il loro tasso di crescita.
Successivamente vengono prescritti radiografie, TAC, spesso PET e a volte RMN, allo scopo di valutare l’estensione della malattia ai vari organi e tessuti. A questi esami si aggiunge la biopsia osteomidollare, utile per determinare la presenza di cellule malate a livello del midollo osseo. Al termine di queste indagini al linfoma viene attribuito lo stadio: da I a IV in base al numero di sedi infiltrate, alla presenza di localizzazioni in organi non linfonodali, e il suffisso B o A alla presenza o meno di segni sistemici (febbre, sudorazione, calo ponderale).
Trattamenti
La scelta della terapia dipende dal tipo e dallo stadio del linfoma non-Hodgkin, dall’età del paziente e dal suo stato di salute generale.
Malattia in stadio iniziale (I-II)
Il paziente affetto da linfoma indolente in stadio iniziale (ossia con malattia localizzata da un lato del diaframma) viene generalmente sottoposto a radioterapia con l’applicazione di radiazioni in dosi adatte per distruggere le cellule neoplastiche. Il paziente affetto da linfoma aggressivo in stadio iniziale, invece, a meno di gravi comorbidità, viene sottoposto ad una breve chemioterapia (3 cicli) associata ad anticorpo monoclonale (rituximab) se la malattia è derivata dai linfociti B, e viene successivamente sottoposto a radioterapia di consolidamento sulla/e sede/i di malattia.
Malattia in stadio avanzato (III-IV)
Il paziente affetto da linfoma indolente in stadio avanzato non sempre necessita di un trattamento immediato: se la malattia non mostra segni clinici di rapida evoluzione, si può rimandarne l’inizio monitorando regolarmente il paziente. In caso contrario vengono effettuati da 6 a 8 cicli chemioterapia, sempre associata a rituximab se il linfoma è di derivazione B-linfocitaria. Il paziente affetto da linfoma aggressivo a uno stadio avanzato viene sottoposto a 6-8 cicli di chemioterapia (sempre associata a rituximab per i linfomi B) da iniziarsi in tempi piuttosto rapidi. Nella scelta della chemioterapia vengono utilizzate combinazioni di farmaci, somministrati tramite iniezione endovenosa, per distruggere le cellule tumorali che crescono rapidamente. Esistono anche farmaci da assumere per via orale, che tuttavia vengono oggi riservati a pazienti anziani o con altre patologie concomitanti che rendono troppo rischiosa la somministrazione della terapia endovenosa. I linfomi aggressivi possono insorgere anche primitivamente in sedi extranodali uniche, ad esempio il cervello, e in questo caso richiedere trattamenti chemioterapici dedicati e radioterapia aggiuntiva.
Radioimmunoterapia
Si tratta di farmaci che combinano un anticorpo monoclonale specifico per il linfoma (di derivazione B linfocitaria) con isotopi radioattivi. Questi composti si legano alle cellule tumorali, tramite l’anticorpo che le riconosce, e successivamente le distruggono attraverso la componente radioattiva. La radioimmunoterapia è generalmente ben tollerata, e gli effetti collaterali sono molto rari, tanto che in alcuni casi viene proposta a pazienti che non possono ricevere terapie aggressive, purché siano monitorati regolarmente per i valori dell’emocromo. In Italia è disponibile un solo tipo di radioimmunoconiugati (ibritumomab tiuxetano: combinazione di anticorpo anti-CD20 rituximab con ittrio90), la cui somministrazione è approvata per i linfomi indolenti follicolari in recidiva oppure in prima linea come consolidamento dopo chemioterapia.
Recidiva
Il trattamento standard della recidiva prevede, in pazienti fino a 65-70 anni di età e in buone condizioni, la chemioterapia ad alte dosi con il trapianto (o meglio il supporto) di cellule staminali autologhe, generalmente periferiche come descritto nella scheda “Linfoma di Hodgkin”. In caso di fallimento anche di questa procedura, viene preso in considerazione il trapianto allogenico di cellule staminali, da fratello/sorella compatibile oppure da donatore volontario.
Leggi anche:
Lo staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o seguici su Twitter, su Instagram o su Pinterest, grazie!
Condividi questo articolo:

 La sclerosi multipla è una malattia mortale?
La sclerosi multipla è una malattia mortale? La progressione da infezione tubercolare a malattia TBC conclamata si verifica quando i bacilli superano le difese del sistema immunitario e iniziano a moltiplicarsi. Nella malattia primaria (1-5% dei casi), questo si verifica subito dopo l’infezione iniziale. Tuttavia, nella maggior parte dei casi, una infezione latente si verifica senza sintomi evidenti. Questi bacilli dormienti producono tubercolosi attiva nel 5-10% dei casi latenti, spesso molti anni dopo l’infezione.
La progressione da infezione tubercolare a malattia TBC conclamata si verifica quando i bacilli superano le difese del sistema immunitario e iniziano a moltiplicarsi. Nella malattia primaria (1-5% dei casi), questo si verifica subito dopo l’infezione iniziale. Tuttavia, nella maggior parte dei casi, una infezione latente si verifica senza sintomi evidenti. Questi bacilli dormienti producono tubercolosi attiva nel 5-10% dei casi latenti, spesso molti anni dopo l’infezione. L’ustione è una lesione dei tessuti tegumentari (pelle ed annessi cutanei) provocata dall’azione di calore, sostanze chimiche, corrente elettrica o radiazioni. Possono essere di varia entità secondo l’intensità della temperatura, la durata del contatto e lo stato fisico della sostanza ustionante (solida, liquida o gassosa); in relazione alla gravità vengono distinte in primo, secondo,terzo e quarto grado.
L’ustione è una lesione dei tessuti tegumentari (pelle ed annessi cutanei) provocata dall’azione di calore, sostanze chimiche, corrente elettrica o radiazioni. Possono essere di varia entità secondo l’intensità della temperatura, la durata del contatto e lo stato fisico della sostanza ustionante (solida, liquida o gassosa); in relazione alla gravità vengono distinte in primo, secondo,terzo e quarto grado.
 Il linfoma di Hodgkin può colpire tutte queste parti del corpo e poi diffondersi ad altri organi. Nel linfoma di Hodgkin, le cellule del sistema linfatico (chiamate linfociti B) crescono in modo anormale e possono accumularsi sia nel sistema linfatico stesso che in altri organi. Con il progredire della malattia, viene compromessa la capacità dell’organismo di combattere le infezioni.
Il linfoma di Hodgkin può colpire tutte queste parti del corpo e poi diffondersi ad altri organi. Nel linfoma di Hodgkin, le cellule del sistema linfatico (chiamate linfociti B) crescono in modo anormale e possono accumularsi sia nel sistema linfatico stesso che in altri organi. Con il progredire della malattia, viene compromessa la capacità dell’organismo di combattere le infezioni.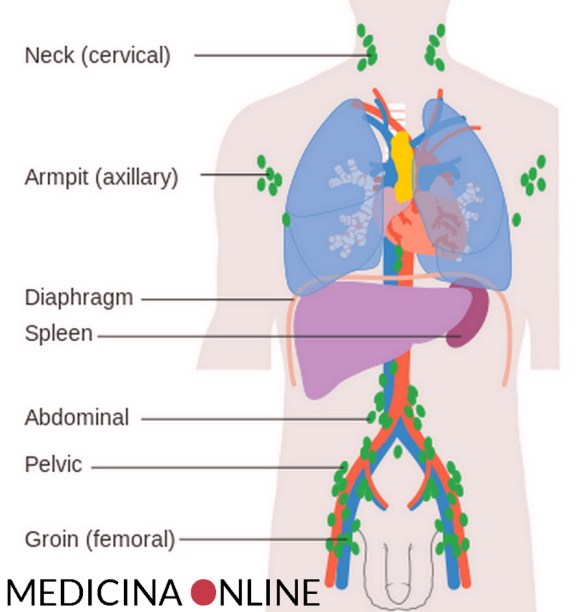 Il linfoma è una malattia del sistema linfatico, cioè dell’insieme di tessuti che hanno la funzione di difendere l’organismo dagli agenti esterni e dalle malattie. Questo sistema comprende i linfonodi, la milza, il timo, il midollo osseo e altre piccole aree dell’organismo. I linfomi fanno parte del più ampio gruppo di neoplasie dei tessuti linfoidi (linfociti T e B e loro precursori). A livello mondiale si stima che attualmente vi siano circa 600.000 linfomi all’anno con circa 300.000 decessi. I linfomi costituiscono il 4% di tutti i tumori e ciò li rende la settima forma più comune tra gli adulti, nei bambini sono il terzo tumore più comune.
Il linfoma è una malattia del sistema linfatico, cioè dell’insieme di tessuti che hanno la funzione di difendere l’organismo dagli agenti esterni e dalle malattie. Questo sistema comprende i linfonodi, la milza, il timo, il midollo osseo e altre piccole aree dell’organismo. I linfomi fanno parte del più ampio gruppo di neoplasie dei tessuti linfoidi (linfociti T e B e loro precursori). A livello mondiale si stima che attualmente vi siano circa 600.000 linfomi all’anno con circa 300.000 decessi. I linfomi costituiscono il 4% di tutti i tumori e ciò li rende la settima forma più comune tra gli adulti, nei bambini sono il terzo tumore più comune.
