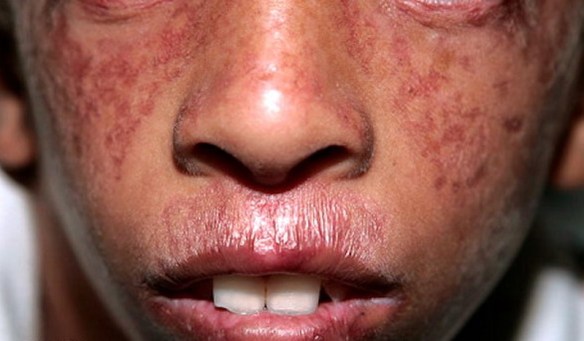
Giovane con eritema teleangiectasico del volto dovuta a sindrome di Bloom
Con “malattia genetica” in medicina si intende un sottotipo di malattia, generalmente rara o molto rara, causata da una o più anomalie del Continua a leggere

Giovane con eritema teleangiectasico del volto dovuta a sindrome di Bloom
Con “malattia genetica” in medicina si intende un sottotipo di malattia, generalmente rara o molto rara, causata da una o più anomalie del Continua a leggere
 Si chiama GENOTIPO l’insieme dei geni che compongono il corredo cromosomico di un organismo. Tenendo in considerazione un solo genotipo, ad esempio quello che determina il colore di un fiore, la proteina che darà il colore del fiore deriva dalla combinazione di due geni, uno sul cromosoma paterno e uno materno. I tipi di alleli di quel gene corrispondono al fenotipo per quel carattere (e per carattere in questo caso indichiamo il colore del fiore).
Si chiama GENOTIPO l’insieme dei geni che compongono il corredo cromosomico di un organismo. Tenendo in considerazione un solo genotipo, ad esempio quello che determina il colore di un fiore, la proteina che darà il colore del fiore deriva dalla combinazione di due geni, uno sul cromosoma paterno e uno materno. I tipi di alleli di quel gene corrispondono al fenotipo per quel carattere (e per carattere in questo caso indichiamo il colore del fiore).
Il FENOTIPO è la manifestazione finale di quel genotipo (nell’esempio precedente: il colore del fiore).
Leggi anche:
Lo staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o seguici su Twitter, su Instagram o su Pinterest, grazie!
 Si dice OMOZIGOTE quando un individuo porta due alleli identici per uno stesso gene (AA o aa), mentre ETEROZIGOTE quando un individuo porta due alleli differenti per lo stesso gene (Aa). Il termine omozigote si riferisce quindi ad un gene in cui l’informazione riportata, che determina il fenotipo, dall’allele materno, è identica a quella paterna. Diversamente, negli eterozigoti, il contributo dell’allele materno e paterno è diverso. In tal caso la determinazione fenotipica è correlata ai concetti di dominanza e recessività genetica.
Si dice OMOZIGOTE quando un individuo porta due alleli identici per uno stesso gene (AA o aa), mentre ETEROZIGOTE quando un individuo porta due alleli differenti per lo stesso gene (Aa). Il termine omozigote si riferisce quindi ad un gene in cui l’informazione riportata, che determina il fenotipo, dall’allele materno, è identica a quella paterna. Diversamente, negli eterozigoti, il contributo dell’allele materno e paterno è diverso. In tal caso la determinazione fenotipica è correlata ai concetti di dominanza e recessività genetica.
Leggi anche:
Lo staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o seguici su Twitter, su Instagram o su Pinterest, grazie!
I geni sono sequenze nucleotidiche situate in posizioni fisse e specifiche del cromosoma. Non sono altro che una serie di nucleotidi con una specifica sequenza che ha la funzione, mediante il successivo processo di TRASCRIZIONE E TRADUZIONE, a generare una proteina (e non solo, in realtà il prodotto può essere anche un tRNA o rRNa ad esempio).
Leggi anche:
Lo staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o seguici su Twitter, su Instagram o su Pinterest, grazie!
La sindrome di Bloom è una malattia autosomica recesssiva caratterizzata da eritema teleangiectasico del volto, fotosensibilita, nanismo ed altre anomalie.
Segni e sintomi
Le tipiche lesioni cutanee sono teleangiectasiche eritematose, appaiono in infanzia al volto, con tipico aspetto a farfalla, ma occasionalmente si riscontrano anche ai dorsi delle mani e dei piedi. Queste aree sono sensibili al sole, e peggiorano dopo l’esposizione. L’ altra caratteristica fondamentale della malattia e la bassa statura, soprattutto prenatale all’esordio, ma che persiste anche in infanzia e in eta adulta. La maggior parte degli individui rimane al di sotto dei 148 cm.
I maschi sono sterili con azospermia, mentre le femmine possono essere fertili.
Altre caratteristiche sono: macchie caffelatte, microcefalia, dolicocefalia, faccia stretta, naso e/o orecchie prominenti, la tipica tonalita alta della voce, l’ipertrigliceridemia. L’intelligenza e normale. Le infezioni sono frequenti, a causa dell’immunodeficienza, e possono essere minacciose per la vita.
Leggi anche:
Storia naturale
La sindrome di Bloom e un disordine genetico che predispone al cancro. Infatti, quasi la meta dei pazienti sviluppano almeno una neoplasia (10% dei quali hanno piu di una neoplasia primitiva, il che e proprio tipico di questa sindrome); l’eta media di insorgenza del primo tumore e di 25 anni (range 2-49 anni). Dopo i trent’anni si manifestano leucemie acute (ALL e ANLL) nel 15% dei casi e linfomi pure nel 15% dei casi. Carcinomi (una grande varieta di tipi) si sviluppano nel 30% dei casi, perlopiu dopo i 20 anni.
A parte le malattie neoplastiche, le complicanze maggiori dal punto di vista medico sono la malattia polmonare cronica e il diabete mellito (nel 10% dei pazienti).
L’incidenza della sindrome e di circa 2/100.000 nati tra gli Ebrei Ashkenazi e tra i Giapponesi (effetto del fondatore: le persone affette derivano da un antenato comune); per il resto e molto rara.
Eziologia
La sindrome di Bloom e trasmessa con modalita autosomica recessiva. Il gene e stato mappato: location 15q26.
Diagnosi
Fa diagnosi il riscontro (patognomonico) di un alto numero di scambi spontanei di cromatidi fratelli (90 per cellula, piu di 10 volte maggiore che nel normale); in alcuni pazienti, il riscontro di una sottopopolazione con basso numero di scambi suggerisce una ricombinazione tra alleli materni e paterni (con mutazioni differenti), che da origine ad un gene selvaggio funzionante. Gli eterozigoti, invece, non si possono investigare con studi citogenetici.
Terapia
Non esistono cure definitive. L’ormone della crescita è controindicato nelle sindromi in cui vi e un aumentato rischio di rotture cromosomiche, come appunto la sindrome di Bloom. Controlli frequenti assicurano la diagnosi precoce dei tumori associati alla sindrome di Bloom. Gli individui affetti da questa patologia devono evitare i raggi X, gli agenti chemioterapici ed altre esposizioni ambientali che possono provocare danni ai loro cromosomi particolarmente fragili.
Leggi anche:
Lo staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o seguici su Twitter, su Instagram o su Pinterest, grazie!
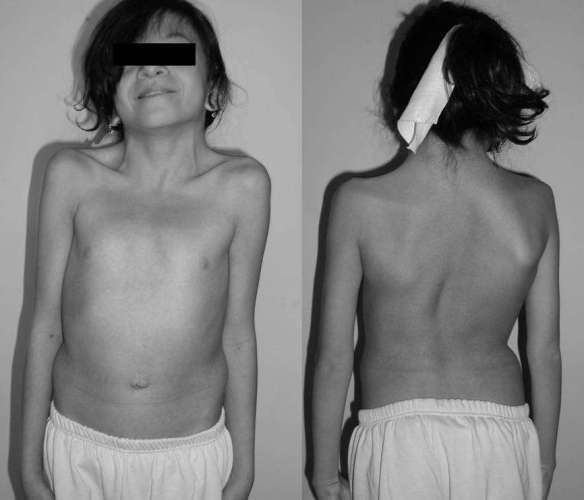 La sindrome di Noonan è una malattia genetica dalle caratteristiche cliniche particolarmente variabili, con un’incidenza stimata di un caso ogni 1000-2500 nati. Le manifestazioni più frequenti includono cardiopatie congenite (nel 70% dei pazienti), bassa statura, un aspetto caratteristico del volto (abbassamento delle palpebre, occhi distanziati, orecchie grandi e ruotate posteriormente), malformazioni del torace, deficit cognitivi variabili, tendenza al sanguinamento e criptorchidismo (mancata discesa dei testicoli nello scroto).
La sindrome di Noonan è una malattia genetica dalle caratteristiche cliniche particolarmente variabili, con un’incidenza stimata di un caso ogni 1000-2500 nati. Le manifestazioni più frequenti includono cardiopatie congenite (nel 70% dei pazienti), bassa statura, un aspetto caratteristico del volto (abbassamento delle palpebre, occhi distanziati, orecchie grandi e ruotate posteriormente), malformazioni del torace, deficit cognitivi variabili, tendenza al sanguinamento e criptorchidismo (mancata discesa dei testicoli nello scroto).
Come si trasmette la sindrome di Noonan?
La sindrome può essere sporadica (il difetto genetico responsabile insorge in modo spontaneo) oppure ereditata dai genitori. In questo caso la trasmissione avviene con modalità autosomica dominante: è sufficiente cioè una copia alterata del gene per sviluppare la malattia. In circa il 50% dei casi, la sindrome è causata da mutazioni del gene PTPN11, mentre in una piccola percentuale di pazienti sono state identificate mutazioni nei geni KRAS, NRAS, RAF1, BRAF e SOS1. Nel complesso, le mutazioni nei geni identificati si riscontrano in circa il 75% dei pazienti con diagnosi clinica della malattia. Mutazioni nei geni SHOC2 e CBL sono state riscontrate in soggetti con caratteristiche cliniche parzialmente sovrapponibili alla sindrome di Noonan.
Leggi anche:
Come avviene la diagnosi della sindrome di Noonan?
La diagnosi avviene in base alle caratteristiche cliniche ed eventualmente all’analisi della storia familiare e può essere confermata con un’indagine genetica (ricerca di mutazioni nei geni PTPN11, RAF1, KRAS, SOS1, NRAS e BRAF). Va comunque ricordato che, per molti pazienti, la causa genetica della malattia non è ancora stata identificata. Inoltre, vista la variabilità clinica della malattia, l’analisi molecolare di altri geni implicati in malattie correlate (HRAS, MEK1, MEK2, CBL e SHOC2) può indirizzare verso una più corretta valutazione clinica. Se una mutazione responsabile è già stata identificata in un componente della famiglia, si può effettuare una diagnosi prenatale con l’indagine genetica del feto attraverso villocentesi o amniocentesi. L’età media alla diagnosi è di nove anni.
Quali sono le possibilità di cura attualmente disponibili per la sindrome di Noonan?
Non esiste una terapia specifica, anche se studi preclinici su modelli animali sono in corso per valutare l’efficacia di alcuni farmaci che inibiscono in maniera specifica l’attività degli enzimi “mutati” responsabili della malattia. Alcune lesioni cardiache devono essere corrette per via chirurgica, mentre in alcuni casi è consigliata l’assunzione di ormone della crescita. In generale, i bambini affetti dalla sindrome vanno tenuti sotto controllo per quanto riguarda la funzionalità cardiaca, la crescita, lo sviluppo motorio, la coagulazione del sangue. Se seguita in modo adeguato, la maggior parte dei bambini colpiti cresce normalmente e raggiunge senza problemi l’età adulta.
Aspettative di vita
La speranza di vita è generalmente normale (paragonabile al resto della popolazione) se sono assenti difetti cardiaci congeniti importanti. L’aspettativa di vita diminuisce all’aumentare della gravità delle patologie cardiache.
Leggi anche:
Lo staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o seguici su Twitter, su Instagram o su Pinterest, grazie!
 Prima di leggere quali sono i fattori di rischio per l’asma bronchiale, ti consiglio di leggere questo articolo: Asma bronchiale in bambini e adulti: cause, sintomi e cura
Prima di leggere quali sono i fattori di rischio per l’asma bronchiale, ti consiglio di leggere questo articolo: Asma bronchiale in bambini e adulti: cause, sintomi e cura
Fattori di rischio ambientali per l’asma bronchiale
Gli allergeni sono considerati un’importante causa di asma bronchiale. L’aumento di incidenza dell’asma riguarda soprattutto le forme ad andamento perenne, in una considerevole parte delle quali è possibile evidenziare una sensibilizzazione ad allergeni indoor, come acari, derivati di animali domestici (gatto e cane) e muffe.
Una meta-analisi sui fattori ambientali considerati responsabili dell’incidenza e della gravità dell’asma, ha concluso che l’esposizione agli allergeni indoor è il fattore ambientale con il più forte effetto sullo sviluppo di asma. Le principali fonti allergeniche degli ambienti esterni sono pollini, derivati da piante erbacee ed arboree e micofiti. Altri agenti responsabili di asma sono i sensibilizzanti professionali. Questi sono responsabili del 9 – 15% dei casi di asma negli adulti. Le sostanze più frequentemente in causa sono isocianati, farina, polvere di cereali e di legno e lattice.
Il fumo di tabacco ha un ruolo importante nello sviluppo di asma ed influenza negativamente il controllo della malattia. L’esposizione al fumo passivo, sia di tipo pre-natale per l’abitudine tabagica della madri durante la gravidanza, sia durante l’infanzia, rappresenta un importante fattore di rischio per lo sviluppo di asma nell’infanzia e nell’età adulta. L’esposizione in età adulta peggiora il controllo dell’asma nelle persone che ne sono affette.
L’esposizione ad inquinanti ambientali è spesso associata a riacutizzazione di un’asma preesistente. Gli inquinanti esterni più comuni sono: ossidi di azoto, ozono, particolato sottile PM10, monossido di carbonio e anidride solforosa. Aumentano prevalentemente durante i mesi invernali nelle città, per il traffico veicolare più frequente, per i riscaldamenti domestici e per le condizioni ambientali climatiche favorevoli alla loro concentrazione. Le costruzioni moderne, caratterizzate da un ridotto ricambio di aria, possono contribuire ad una maggior esposizione ad inquinanti chimici (fumi e vapori irritanti) presenti negli ambienti interni (indoor) derivanti dalla combustione del gas e dai detersivi. Anche le infezioni virali delle vie aeree sono state associate allo sviluppo di asma. Se contratte nella prima infanzia, come nel caso delle infezioni da virus respiratorio sinciziale (RSV), causano frequentemente wheezing e bronchiolite, che nel corso degli anni diventano un fattore favorente lo sviluppo di asma non allergica. Infezioni virali in età adulta possono anche slatentizzare una reattività bronchiale misconosciuta e rappresentare l’esordio dell’asma.
Esistono inoltre alcune condizioni patologiche che possono facilitare l’insorgenza di asma o favorirne le riacutizzazioni. Poliposi nasale, rinite, rino-sinusite, reflusso gastroesofageo possono contribuire alla manifestazione dell’asma. Il controllo di queste malattie, pertanto, favorisce anche il controllo dell’asma, riducendo la frequenza delle riacutizzazioni.
Fattori di rischio genetici per l’asma bronchiale
L’atopia è una predisposizione geneticamente determinata a produrre un eccesso di IgE in risposta all’esposizione ad allergeni, e si evidenzia con la dimostrazione di aumentati livelli sierici di IgE specifici e/o con una risposta positiva ai test allergometrici cutanei (prik test) effettuati con una batteria di allergeni inalanti standardizzati. La proporzione di asme attribuibili all’atopia è circa la metà dei casi. L’atopia presenta una familiarità; pertanto, si apprezza un aumento del rischio di sviluppare l’asma in presenza di genitori atopici affetti da asma. La manifestazione dell’atopia ha una storia naturale: di solito la dermatite atopica precede lo sviluppo di rinite allergica e di asma. La rinite allergica rappresenta quindi un importante fattore di rischio per lo sviluppo di asma. Non a caso, spesso le due patologie coesistono nello stesso paziente e in molti casi la rinite allergica precede lo sviluppo di asma. Altro elemento da considerare è l’eventuale presenza di wheezing (sibilo che caratterizza il respiro del neonato) ricorrente nei primi anni di vita. Una parte di questi bambini svilupperà asma.
Lo Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o unisciti al nostro gruppo Facebook o ancora seguici su Twitter, su Instagram o su Pinterest, grazie!

La personalizzazione del trattamento per ciascun paziente con tumore del colon-retto con metastasi ha l’obiettivo di ottenere il massimo vantaggio, in termini di efficacia, dalla somministrazione dei farmaci a bersaglio. A tale scopo il medico che ha in cura il malato può richiedere un esame di laboratorio chiamato esame (test) del RAS. Questo esame definisce particolari caratteristiche genetiche delle cellule tumorali. Quando nelle cellule si verifica una variazione dei geni, si parla di mutazione. I geni indicati con l’acronimo RAS, includono il KRAS e il NRAS. I geni RAS funzionano come “interruttori” che attivano i meccanismi di crescita e replicazione delle cellule tumorali e possono essere nello stato normale (definito in inglese wild-type: “tipo selvaggio”) o mutato (alterato) . Il test RAS identifica i pazienti con tumore del colon-retto metastatico nei quali una particolare terapia biologica, con anticorpi monoclonali anti-EGFR, è più efficace in base alla presenza di geni RAS normali o mutati.
La risposta dei singoli pazienti ai trattamenti antitumorali è variabile. Non tutti i pazienti hanno lo stesso beneficio dallo stesso farmaco. Questo avviene sia per la chemioterapia, che per le terapie biologiche a bersaglio molecolare. Tuttavia, la ricerca scientifica sta progressivamente evidenziando particolari caratteristiche, proprie del paziente o del tumore, che permettono di prevedere chi ha più probabilità di giovarsi di un determinato trattamento. Tali caratteristiche corrispondono a variabili di laboratorio definite “biomarcatori”. Grazie alle informazioni fornite dai biomarcatori, i medici sono in grado di personalizzare la terapia e di aumentare le probabilità di successo, nei soggetti sensibili al trattamento. Per i pazienti con tumore del colon-retto metastatico i biomarcatori sono le mutazioni dei geni RAS (KRAS e NRAS).
Leggi anche:
Il test dei RAS solitamente non richiede procedure invasive aggiuntive, in quanto viene eseguito sui campioni di tessuto (biopsie) raccolti nel corso della valutazione della malattia. L’esame dei RAS, consistente in un’analisi di tipo genetico, viene eseguito su una piccola quantità di materiale bioptico prelevato dal tumore primario o dalle metastasi. Il risultato dell’esame dei RAS, normalmente disponibile in circa una decina di giorni, valuta i geni KRAS e NRAS, indicando se tali geni sono presenti allo stato normale (o wild-type) o mutato. Lo stato normale dei geni KRAS e NRAS indica che il paziente ha maggiori probabilità di rispondere a una terapia a base di anticorpi monoclonali anti-EGFR, mentre nei casi con geni KRAS e NRAS mutati la somministrazione del farmaco non è indicata perché non efficace.
KRAS e NRAS sono i nomi, sia dei geni RAS, che delle proteine che essi codificano. Le proteine RAS giocano un ruolo importante nel processo di crescita e moltiplicazione cellulare, infatti, agiscono come “interruttori” che servono ad “accendere”, a seguito della interazione fra EGF e recettore, la moltiplicazione cellulare. Nelle cellule normali e in quelle del tumore del colon-retto metastatico senza mutazioni dei geni RAS, tali interruttori si accendono e poi si spengono immediatamente, in un’alternanza che attiva o disattiva, appunto, la replicazione cellulare. Se uno dei geni RAS non è mutato, gli anticorpi che bloccano il recettore dell’EGF (anti-EGFR) si sono dimostrati un valido strumento terapeutico per contrastare il tumore del colon-retto metastatico. Se invece il gene RAS è mutato, cioè è modificato nella sua composizione, anche la proteina da esso prodotta sarà diversa, e si comporterà come un interruttore perennemente “acceso”, indipendentemente dagli stimoli esercitati dal fattore di crescita EGF. Le mutazioni dei geni RAS sono presenti circa nel 45% dei tumori del colon-retto metastatici.
I biomarcatori sono estremamente importanti nel trattamento dei tumori, in quanto aiutano i medici a scegliere i trattamenti che presumibilmente saranno più efficaci per ciascun paziente. In particolare l’esame di geni RAS si esegue nei laboratori di anatomia patologica e biologia molecolare dei principali ospedali italiani.
Leggi anche:
Lo Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o unisciti al nostro gruppo Facebook o ancora seguici su Twitter, su Instagram o su Pinterest, grazie!
