
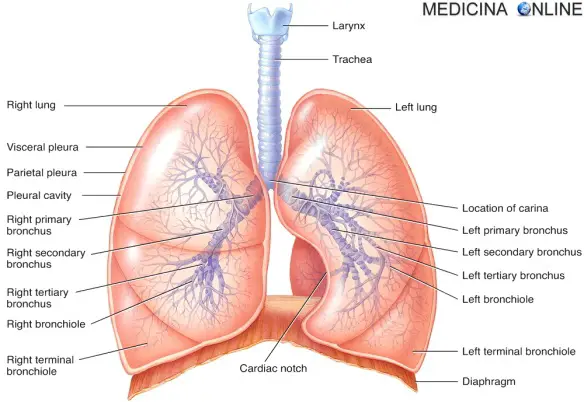
 La discinesia ciliare primaria (anche chiamata “discinesia ciliare primitiva” o “sindrome di Siewert” o “sindrome di Siewert-Kartagener”) da cui l’acronimo “DCP”, una volta chiamata “sindrome delle ciglia immobili“, in inglese “primary ciliary dyskinesia” (da cui l’acronimo “PCD”) o “immotile-cilia syndrome” o “Siewert’s syndrome” o “Siewert-Kartagener syndrome”, è un gruppo di rare patologie congenite a trasmissione autosomico recessiva (anche se recentemente sono state scoperte anche forme a trasmissione autosomico dominante) dovute ad alterazioni della struttura e/o della funzione delle ciglia mobili di diversi tessuti. La discinesia può interessare varie strutture anatomiche, tuttavia interessa soprattutto le ciglia degli epiteliociti della mucosa respiratoria con conseguente difetto del trasporto mucociliare che causa un’elevata morbilità delle alte e basse vie aeree, determinando broncorrea cronica con bronchiectasia e sinusite cronica.
La discinesia ciliare primaria (anche chiamata “discinesia ciliare primitiva” o “sindrome di Siewert” o “sindrome di Siewert-Kartagener”) da cui l’acronimo “DCP”, una volta chiamata “sindrome delle ciglia immobili“, in inglese “primary ciliary dyskinesia” (da cui l’acronimo “PCD”) o “immotile-cilia syndrome” o “Siewert’s syndrome” o “Siewert-Kartagener syndrome”, è un gruppo di rare patologie congenite a trasmissione autosomico recessiva (anche se recentemente sono state scoperte anche forme a trasmissione autosomico dominante) dovute ad alterazioni della struttura e/o della funzione delle ciglia mobili di diversi tessuti. La discinesia può interessare varie strutture anatomiche, tuttavia interessa soprattutto le ciglia degli epiteliociti della mucosa respiratoria con conseguente difetto del trasporto mucociliare che causa un’elevata morbilità delle alte e basse vie aeree, determinando broncorrea cronica con bronchiectasia e sinusite cronica.
Se la discinesia ciliare primaria si associa a situs inversus con destrocardia (cuore con apice diretto a destra e fegato a sinistra), allora si parla di “sindrome di Kartagener“.
Cenni storici
La classica combinazione di sintomi associata alla discinesia ciliare primaria fu descritta per la prima volta nel 1904 da AK Siewert, mentre Manes Kartagener pubblicò il suo primo rapporto sull’argomento nel 1933; il disturbo è, per quarto motivo, spesso indicato come sindrome di Siewert o sindrome di Siewert-Kartagener. Il termine “sindrome delle ciglia immobili” è attualmente in disuso, dal momento che non descrive ottimamente la malattia visto che le ciglia non sono necessariamente immobili bensì presentano un certo grado di movimento a seconda della mutazione genetica coinvolta.
Epidemiologia
La discinesia ciliare primaria ha una prevalenza stimata in 1/20.000. Pur essendo rara è comunque, per frequenza, la seconda malattia congenita dell’apparato respiratorio dopo la fibrosi cistica.
Età di esordio
La discinesia ciliare primaria è una condizione congenita, cioè già presente alla nascita; i sintomi generalmente esordiscono nel periodo neonatale, cioè subito dopo la nascita.
Cause e geni coinvolti
La discinesia ciliare primaria ha cause genetiche ed è a trasmissione mendeliana, autosomico recessiva; inoltre è una malattia congenita (già presente alla nascita) e geneticamente eterogenea (possono esserci diverse mutazioni in individui diversi che possono portare alla stessa patologia). La ragione della eterogeneità genetica è da ricercare nella molteplicità di proteine che compongono un ciglio mobile (circa 250). Circa il 90% degli individui affetti da DCP possiede mutazioni a carico dei bracci interni/esterni di dineina. Le proteine che compongono questi bracci sono situate in loci diversi dell’organismo umano ed ecco perché i sintomi possono variare a seconda dell’individuo. Sono state descritte diverse anche altre anomalie strutturali come il deficit del raggio radiale o la trasposizione dei microtubuli periferici. La malattia è dovuta alle mutazioni dei geni DNAH5 (circa il 30% dei pazienti), DNAI1 (2-10% dei pazienti), TXNDC3, DNAH11, e DNAI2 (identificati più raramente), geni che codificano per le componenti del braccio esterno della dineina (ODA), che generano il movimento dei flagelli e delle ciglia. Le mutazioni dei geni DNAI1 o DNAH5 causano circa un terzo dei casi di DCP/SK. Recentemente sono stati associati alla DCP altri geni: C14orf104, OFD1, RSPH9, e RSPH4A. Una forma rara di DCP associata alla retinite pigmentosa è dovuta a una mutazione del gene RPGR.
IMPORTANTE: recentemente sono state descritte alcune famiglie nelle quali la trasmissione della discinesia ciliare primaria era autosomica dominante, anche se non sono stati identificati i geni coinvolti.
Trasmissione autosomica recessiva
Una malattia è detta a trasmissione autosomica recessiva quando l’allele alterato deve essere presente in coppia (omozigosi), cioè sono necessarie due copie dell’allele difettoso per far sì che la malattia si esprima, a prescindere dal sesso. Non basta un solo genitore portatore sano o malato, bensì entrambi i genitori devono essere portatori sani o malati. Il fenotipo quindi si esprime quando nel genotipo dell’individuo sono presenti entrambi gli alleli responsabili, fatto che spiega l’alta probabilità di sviluppare malattie genetiche in caso di incesto. Quindi:
- un individuo che possegga entrambi gli alleli alterati: è portatore ed è malato;
- un individuo che possegga solo un allele alterato: è portatore ma è sano;
- un individuo che non possegga nessun allele alterato: NON è portatore ed è sano.
Essere portatore sano vuol dire quindi NON avere la patologia ma possedere nel proprio genotipo un allele mutato, che può essere trasmesso alle generazioni successive.
Dalla combinazione delle possibili condizioni di genitori sani, malati e portatori sani, deriva la distribuzione probabilità che la malattia sia trasmessa ai figli:
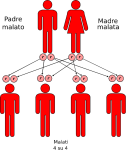
- genitori malato-malato: la probabilità che il figlio/a nasca malato è del 100%;
- genitori sano-malato: la probabilità che il figlio/a nasca portatore sano è del 100%;
- genitori malato-portatore sano: la probabilità che il figlio/a nasca malato è del 50% e del 50% che nasca portatore sano;
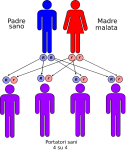
- genitori sano-portatore sano: la probabilità che il figlio/a nasca sano è del 50% e del 50% che nasca portatore sano;
- genitori portatore-portatore: la probabilità che il figlio/a nasca portatore sano è del 50% mentre è del 25% che nasca sano o malato.
Se nessuno dei genitori ha un allele mutato, non c’è ovviamente alcuna trasmissione autosomica recessiva ed i figli saranno tutti sani e NON portatori dell’allele mutato.
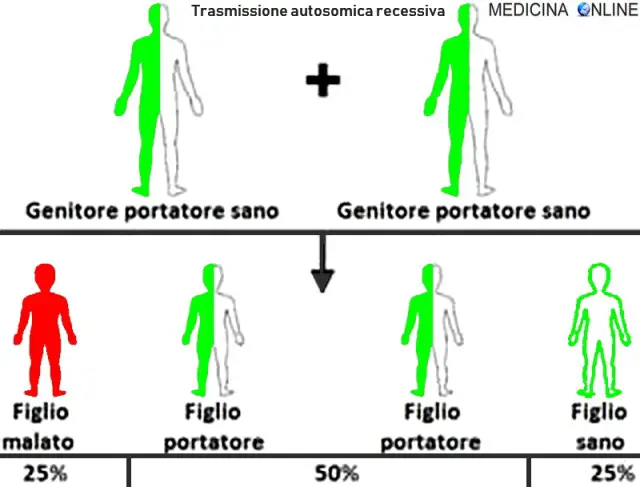
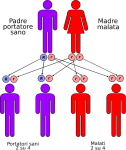
Nell’immagine che segue, è raffigurata la tipica situazione in cui entrambi i genitori sono sani ma portatori dell’allele mutato:
- un figlio su quattro avrà entrambi gli alleli alterati e sarà malato ed ovviamente portatore;
- due figli su quattro avranno un allele normale ed uno alterato e saranno sani ma anche portatori;
- un figlio su quattro avrà entrambi gli alleli normali e sarà sano e NON portatore.
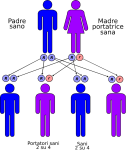
Le altre quattro situazioni possibili sono raffigurate nelle seguenti immagini:
Sintomi e segni
I segni clinici possono essere presenti alla nascita in associazione con una sindrome da insufficienza respiratoria. Durante l’infanzia, la malattia si presenta con tosse e broncorrea cronica, rinite cronica associata a poliposi nasale, agenesia del seno frontale e otite ricorrente. L’associazione con il situs inversus è presente in quasi il 50% dei casi e corrisponde alla sindrome di Kartagener. I difetti ciliari provocano un’alterazione del trasporto mucociliare nell’apparato respiratorio. Questi difetti possono colpire anche i flagelli degli spermatozoi, rendendo i maschi sterili. Possono essere presenti cardiopatie congenite, compreso il difetto del setto atriale o la trasposizione dei grossi vasi. Per approfondire:
- Difetti del setto interatriale: cause, sintomi, diagnosi, terapia, rischi, prognosi, mortalità
- Trasposizione completa e corretta delle grandi arterie, cuore univentricolare e intervento di Fontain
Diagnosi
Sono stati proposti diversi test diagnostici per questa condizione. Questi includono i livelli nasali di ossido nitrico come test di screening, la microscopia ottica di biopsie per il pattern del battito ciliare e la frequenza e l’esame al microscopio elettronico dei geni interessati, come metodo diagnostico definitivo. Sono stati proposti anche test genetici, ma questo è difficile dato che sono coinvolti più geni. La sterilità è uno dei segni diagnostici più suggestivi nei maschi all’anamnesi. Il test da inalazione di saccarina può fornire informazioni utili, anche se non è adatto ai bambini piccoli, in quanto il paziente deve rimanere fermo durante il test. Di conseguenza, per facilitare la diagnosi è di solito necessaria la biopsia o il ‘brushing’ della mucosa ciliare, che dovrebbe essere effettuata presso centri specializzati. La biopsia consente di valutare la funzione ciliare attraverso le misurazioni della frequenza del battito ciliare e di analizzare la struttura ciliare al microscopio elettronico. È possibile la registrazione del battito ciliare con una videocamera digitale ad alta velocità in slow motion.
Diagnosi differenziale
La diagnosi differenziale si pone con la fibrosi cistica e l’immunodeficienza. Per approfondire:
- Fibrosi cistica: storia, organi coinvolti, cause, trasmissione, anatomia patologica
- Fibrosi cistica polmonare: cos’è, sintomi in neonati e bambini, cureFibrosi cistica: sintomi, segni, diagnosi, esami
- Fibrosi cistica: terapia farmacologica e chirurgica, prognosi, mortalità
Terapia
Non ci sono strategie di trattamento efficaci standardizzate per la discinesia ciliare primaria. Le attuali terapie sono estrapolate dalla fibrosi cistica e dai pazienti con bronchiectasie non FC e mancano di convalida per l’uso specifico della discinesia ciliare primaria. La terapia è sintomatica. Le vaccinazioni regolari e i trattamenti antibiotici precoci, associati alla fisioterapia respiratoria, in caso di infezioni polmonari, aiutano a limitare lo sviluppo della bronchiectasia. Può svilupparsi una grave insufficienza respiratoria fatale; il trattamento a lungo termine con macrolidi come claritromicina, eritromicina e azitromicina è stato applicato empiricamente per il trattamento della discinesia ciliare primaria in Giappone, sebbene controverso a causa degli effetti dei farmaci. È necessaria una fisioterapia polmonare giornaliera per i pazienti affetti da bronchiectasia, anche durante i periodi di remissione. Nei pazienti affetti da insufficienza respiratoria allo stadio finale, deve essere proposto il trapianto polmonare. La chirurgia si rende a volte necessaria per la correzione delle cardiopatie.
Prognosi
La prognosi della malattia dipende dalla ricorrenza delle infezioni polmonari, dato che molto spesso esitano in bronchiectasie associate a insufficienza respiratoria cronica. Non esiste, tuttavia, una stima affidabile dell’aspettativa di vita per le persone con discinesia ciliare primaria. Il più grande studio multicentrico sulla funzione polmonare nelle persone con discinesia ciliare primaria in molti paesi europei ha trovato forti prove che confutano un presupposto comune che si tratti di una malattia lieve. Questo studio ha scoperto che la funzione polmonare delle persone con discinesia ciliare primaria è paragonabile a quella di quelle con fibrosi cistica nell’infanzia, ma è migliore nella giovane età adulta. Entrambe le malattie, tuttavia, sono progressive e la funzione polmonare diminuisce con l’età rispetto ai gruppi di coetanei. I dati dell’indagine indicano che i sintomi respiratori aumentano progressivamente e continuamente a partire dalla metà degli anni ’20 rispetto alla norma della popolazione.
Per approfondire:
- Sindrome di Kartagener: cause, sintomi, diagnosi, terapia e prognosi
- Situs solitus, situs inversus, situs viscerum specularis, situs viscerum inversus totalis e situs ambiguous
- Destrocardia, dexiocardia, cuore speculare, destroversione e destroposizione
- Eterotassia (eterotaxia): etimologia, tipi, gravità, cause, sintomi, diagnosi e terapia
- Ectopia cordis: tipi, classificazione, cause, malformazioni associate, prognosi
Leggi anche:
- Com’è fatto il cuore, a che serve e come funziona?
- Cosa si trova a sinistra e a destra del corpo umano?
- Perché il cuore si trova a sinistra e non a destra nel torace?
- Stenosi aortica congenita e valvola aortica bicuspide
- Stenosi congenita della valvola polmonare e anomalia di Ebstein
- Sindrome della scimitarra: cause, sintomi, diagnosi, terapie, prognosi e mortalità
- Difetto del setto interventricolare: cause, sintomi, diagnosi, rischi, terapie, prognosi
- Difetti del setto interventricolare: cause, sintomi, diagnosi e terapie
- Sindrome di Eisenmenger: diffusione, cause, sintomi, segni, diagnosi, terapia, attività sportiva
- Tetralogia di Fallot: caratteristiche, diffusione, comorbilità, etimologia e cenni storici
- Coartazione aortica: cause, sintomi, diagnosi, terapie complicanze e rischi
- Dotto arterioso pervio: cause, sintomi, diagnosi, terapie complicanze e rischi
- Tetralogia di Fallot: cause, sintomi, diagnosi, terapie complicanze e rischi
- Differenza tra difetto del setto interatriale, interventricolare e dotto arterioso pervio SCHEMA
- Ipertensione polmonare persistente nel neonato: trattamento, prognosi, mortalità
- Pervietà del dotto di Botallo: cause, sintomi, diagnosi, terapia, prognosi, rischi nell’adulto
- Forame ovale pervio: cause, sintomi, diagnosi, rischi, terapie, prognosi
- Come si muove il sangue all’interno del cuore?
- Differenza tra circolazione sistemica, polmonare ed intracardiaca
- Come si muove l’impulso elettrico cardiaco nel cuore?
- Elettrocardiogramma (ECG) a riposo e sotto sforzo: cos’è ed a che serve?
- ECG: cosa indicano le onde P, T, U, il complesso QRS ed il segmento ST
- Ritmo sinusale ECG: normofrequente, tachicardico, valori, ai limiti della norma
- Sindrome del QT lungo: valorie, cause, cura, farmaci, sportivi
- Effetto Doppler: cos’è e come viene usato in campo medico?
- Ecocolordoppler: cos’è, a che serve e come funziona?
- Ecocolordoppler arterioso e venoso degli arti inferiori e superiori
- Ecocolordoppler cardiaco (ecocardio): funzioni, preparazione, gravidanza
- Ecocardiogramma per via transesofagea: preparazione, è doloroso?
- Differenza tra ecocardiografia ed elettrocardiogramma
- Ecocardiogramma da stress (ecostress) fisico e farmacologico: come si svolge, è pericoloso?
- Endocardite batterica: profilassi in bambini ed adulti
- Valvole cardiache: cosa sono, quali sono ed a che servono?
- Insufficienza della valvola mitralica lieve, moderata, severa: sintomi, diagnosi e terapia
- Insufficienza mitralica lieve, moderata, severa e sport
- Insufficienza polmonare lieve, severa, acuta: sintomi e cura
- Prolasso mitralico: gravità, sintomi, sport, ansia, intervento minivasivo
- Insufficienza aortica: lieve, sport, pressione differenziale, acuta
- Insufficienza tricuspidale: lieve, severa, sport, soffio, sintomi
- Stenosi aortica: lieve, severa, sintomi, intervento, nell’anziano
- Stenosi mitralica: cause, sintomi, diagnosi, esami, rischi, complicanze, farmaci, terapie chirurgiche
- Stenosi e insufficienza tricuspidale: cause, sintomi, diagnosi e terapia
- Stenosi polmonare, insufficienza polmonare e patologia multivalvolare
- Cardiopatia reumatica, valvole cardiache protesiche e profilassi per l’endocardite
- Differenza tra prolasso e insufficienza mitralica
- Differenza tra insufficienza e stenosi valvolare
- Mediastino: anatomia, suddivisione, inferiore, anteriore, superiore
- Sindrome mediastinica: cause, sintomi e cura delle malattie del mediastino
- Tumori del mediastino: timomi e neurinomi, sintomi e cure
- Mediastinite acuta, cronica e fulminante: diagnosi, sintomi e cura
- Massa mediastinica: sintomi, cause, localizzazione, terapie
- Cardiomiopatia dilatativa: cause, sintomi, diagnosi e terapia
- Cardiomiopatia ipertrofica: cause, sintomi, diagnosi, terapia e prognosi
- Cardiomiopatia restrittiva: classificazione, cause, sintomi, diagnosi e terapia
- Cardiomiopatia alcolica e cardiomiopatia ventricolare destra aritmogena
- Miocardite: cause, sintomi, diagnosi e terapia
- Miocardite: terapia, conseguenze, recupero, morte
- Pericardite acuta: cause, sintomi, diagnosi e terapia
- Pericardite fulminante e cronica: ECG, cura, contagio
- Pericardite costrittiva e pericardite essudativa costrittiva
- Versamento pericardico: cause, sintomi, diagnosi e terapia
- Versamento pericardico lieve moderato severo: cura e riassorbimento
- Tamponamento cardiaco: cause, sintomi, diagnosi e terapia
- Tamponamento cardiaco: sintomi, ECG, polso paradosso, linee guida
- Cardiomegalia: sintomi, congenita, cura, diagnosi con RX
- Traumi del torace: epidemiologia, cause e cenni di medicina legale
- Fisiopatologia dei traumi toracici: lesioni della parete toracica, polmonari e delle vie aeree
- Fisiopatologia dei traumi toracici: lesioni del cuore, dei grossi vasi e del diaframma
- Traumi del torace: aspetti clinici, terapia, assistenza alle vie aeree e ventilatoria
- Morte cardiaca improvvisa: cause, sintomi premonitori e cure
- Rottura di cuore: cos’è, sintomi, terapia
- Aritmia cardiaca: cause e fattori di rischio, sintomi, diagnosi e cura
- Anatomia macroscopica del cuore, sistema circolatorio e circolazione coronarica
- Sistema di conduzione dell’impulso elettrico nel cuore e innervazione del miocardio
- Miocardio, miofibrilla, sarcomero e contrazione muscolare calcio-dipendente
- Fisiologia della circolazione: ciclo cardiaco e performance cardiaca
- Fisiologia della circolazione coronarica, sistemica e polmonare
- Risposta cardiovascolare allo sforzo ed all’esercizio fisico
- Di cosa è composto il sangue e quali sono le sue funzioni?
- Saturazione dell’ossigeno: valori normali e patologici in anziani e bambini
- Circolazione coronarica, arterie e vene coronarie: anatomia e funzioni
- Riduzione della riserva coronarica: cos’è e come si studia
- Gruppi sanguigni: cosa sono e quali sono compatibili tra loro
- Cardiopatia ischemica: cronica, definizione, sintomi, conseguenze
- Infarto cardiaco: sintomi premonitori, cause, cosa fare, enzimi, cure
- Sindrome coronarica acuta: sintomi, terapia, classificazione, cura
- Insufficienza cardiaca (scompenso cardiaco): cause, sintomi iniziali, tipi, cure
- Insufficienza cardiaca: sintomi iniziali, sinistra, acuta, cronica
- Angina pectoris stabile, instabile, secondaria: sintomi, interpretazione e terapia
- Palpitazioni (cardiopalmo) a riposo, da ansia, notturne, dopo mangiato
- Differenza tra ipossiemia e ipercapnia
- Differenza tra ipossiemia, ipossia, anossiemia ed anossia
- Ipossia: valori, conseguenze, sintomi, cure
- Ipossiemia: significato, valori, sintomi, conseguenze, rischi, cure
- Ipercapnia: valori, terapia, conseguenze e trattamento
- Anossia: definizione, cause, sintomi, sinonimo, cure
- Ipocapnia: significato, cause, valori, alcalosi respiratoria
- Ossigenoterapia: uso, controindicazioni, domiciliare, con maschera
- Ipertensione polmonare: lieve, severa, terapia, aspettativa di vita
- Shunt in medicina: cardiaco, cerebrale, polmonare ed altre tipologie
- Pentalogia di Cantrell: il cuore batte fuori dal corpo [VIDEO]
- Sindrome di Wolff-Parkinson-White: cos’è, cosa fare, come si cura
- Bimba nata con organi fuori dal corpo, glieli avvolgono con una pellicola
- Il miracolo della bambina nata con mezzo cuore
- Anastomosi vascolare arteriosa e venosa: tipi e differenze
- Malattie cardiovascolari: i 10 comandamenti del cuore in salute
- Plasma e cellule (elementi corpuscolati) che compongono il sangue
- Plasma e suoi derivati (plasmaderivati)
- Differenza tra sangue, plasma e siero
- Come si ottiene il plasma?
- Pugno precordiale sul petto: significato, quando farlo, linee guida
- Defibrillatore: cos’è, come funziona, prezzo, voltaggio, manuale ed esterno
- Bradicardia: sintomi, conseguenze, rimedi, notturna e grave
- Fibrillazione atriale: terapia, rischi, cosa fare, ECG, quando preoccuparsi
- Differenza tra fibrillazione ventricolare ed arresto cardiaco
- Arresto cardiaco: conseguenze, cause, coma, terapia, cosa fare
- Fibrillazione ventricolare: cos’è, terapia, cause scatenanti, frequenza
- Tachicardia improvvisa: cosa fare, ansia, rimedi, valori, dopo i pasti
- Extrasistole: a riposo, ansia, sono pericolose, cure e gravidanza
- Fibrillazione atriale: farmaci e terapia dell’aritmia cardiaca
- Farmaci antiaritmici: meccanismo d’azione ed effetti collaterali
- Primo soccorso e BLS (Basic Life Support): cos’è e come si fa
- Massaggio cardiaco: quando farlo e come farlo [LINEE GUIDA]
- Enzimi cardiaci: alti, bassi, tempi, risultati, ogni quante ore
- Coronarografia: preparazione, stent, angioplastica, è pericolosa
- Angioplastica coronarica: convalescenza, dieta, stent, dolori, durata
- Holter cardiaco (ECG dinamico) 24 ore: lettura risultati, valori, costo
- Radiografia del torace: come si fa, indicazioni, bisogna spogliarsi, costo
- Farmaci trombolitici (fibrinolitici): nomi commerciali e indicazioni
- Antiaggreganti piastrinici: nomi commerciali, effetti collaterali
- Farmaci anticoagulanti : elenco ed effetti collaterali
- Cardiopatia ischemica: cronica, definizione, sintomi, conseguenze
- Infarto, ischemia, necrosi, aterosclerosi, trombo, embolo, ictus, miocardio… Facciamo chiarezza
- Rene ectopico ed ectopia renale: tipi, sintomi, diagnosi e cure
- Ectopia: cosa significa, differenti tipologie, sintomi e cure
- Si può vivere senza reni? Conseguenze della nefrectomia
- Differenza tra topico ed ectopico in medicina
Dott. Emilio Alessio Loiacono
Medico Chirurgo
Direttore dello Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o unisciti al nostro gruppo Facebook o ancora seguici su Twitter, su Instagram, su YouTube, su LinkedIn, su Tumblr e su Pinterest, grazie!
